Прогнозный экономический эффект применения алирокумаба у пациентов с гиперхолестеринемией и высоким сердечно-сосудистым риском
Оценка экономической целесообразности включения алирокумаба в государственные программы возмещения для льготных категорий граждан.
На основании результатов опубликованных исследований проведена сравнительная оценка влияния на бюджет медицинских технологий с добавлением алирокумаба к терапии статинами и эзетимибом и стандартной терапии статинами и эзетимибом для вторичной профилактики после острого коронарного синдрома (ОКС) с горизонтом времени 2 года. Использовали Марковское моделирование и расчет прямых медицинских затрат. Для проверки устойчивости результатов проведен анализ чувствительности в отношении изменения стоимости алирокумаба. Стоимость болезни рассчитывали как совокупность прямых медицинских затрат (препараты, лечение сердечно-сосудистых заболеваний, аферез) и непрямых затрат (потери валового внутреннего продукта вследствие временной нетрудоспособности, сердечно-сосудистого заболевания или смертельного исхода).
Включение алирокумаба в программу государственного возмещения позволяет уменьшить нагрузку на бюджет на 49,05% у больных высокого сердечно-сосудистого риска, в том числе у больных семейной гиперхолестеринемией и рефрактерностью к стандартной гиполипидемической терапии. Экономия возникает в том числе за счет меньшей потребности в аферезе липидов плазмы крови при применении алирокумаба. Уменьшение финансового бремени болезни более чем на 50% в расчете на трудоспособное население при использовании алирокумаба ожидается за 2 года. Анализ чувствительности подтвердил устойчивость полученных результатов при искусственном увеличении цены препарата в 2,84 раза. Применение алирокумаба позволяет снизить бремя заболевания на 56,49%, или 7,78 млрд руб., в год для целевой группы за 2 года терапии по сравнению с текущей практикой лечения (с учетом непрямых затрат).
Полученные результаты позволяют считать включение алирокумаба в программы государственного (страхового) возмещения для вторичной профилактики после перенесенного ОКС экономически оправданным.
Многочисленные популяционные исследования свидетельствуют о том, что при применении гиполипидемических препаратов в реальной клинической практике необходимый (безопасный) уровень липидов крови не достигается у большинства пациентов [1-3]. Особое внимание уделяется тому факту, что даже у пациентов группы самого высокого риска, у которых должна проводиться наиболее «жесткая» вторичная профилактика, например, после острого коронарного синдрома (ОКС), стандартная гиполипидемическая терапия часто не обеспечивает целевой уровень холестерина липопротеидов низкой плотности (ЛНП) [4]. Как подчеркивают исследователи, одной из причин неэффективности вторичной профилактики статинами является возможная непереносимость высоких доз этих препаратов. Так, в клинических исследованиях частота миопатии, вызванной статинами, достигала 5%, а в обычной клинической практике это осложнение при использовании оптимальных доз статинов встречалось еще чаще [5,6]. Снижению эффективности вторичной профилактики сердечнососудистых событий, включая преждевременную смерть, может способствовать невыполнение больными врачебных рекомендаций, касающихся терапии статинами [7]. Более половины пациентов, нуждающихся в статинах, в первую очередь для профилактики повторных событий, получают субоптимальные дозы или вообще не принимают эти препараты [8], а среди оставшихся велика доля пациентов с высоким риском сердечно-сосудистой смерти (SCORE>20%) и низкой приверженностью к терапии статинами [9].
Не лучше обстоят дела с приемом гиполипидемических препаратов и у больных, перенесших первый ОКС. Перед выпиской из стационара таким пациентам практически всегда рекомендуют лечение статинами, однако через несколько лет эти препараты продолжают принимать не более 41% больных, да и то в низких, а, следовательно, малоэффективных, суточных дозах [10]. В европейских странах в течение года после выписки из стационара 40–75% пациентов, перенесших ОКС, также перестают принимать статины [11]. На приверженность к лечению влияют различные факторы, среди которых центральную роль играет поведение больного, который, сталкиваясь с побочными эффектами статинов или опасаясь их потенциальной токсичности, прекращает лечение или самостоятельно снижает дозу препарата. Опрос врачей свидетельствует о том, что и специалисты подвержены определенной инерции в принятии решения о терапии и нередко рекомендуют дозы, явно недостаточные для достижения целевого уровня липидов [12].
Особое внимание следует уделять больным сахарным диабетом 2 типа (СД), у которых чаще развиваются тяжелые сердечно-сосудистые события [13]. Так, вероятность ишемической болезни сердца (ИБС) у больных СД 2 типа по сравнению с другими категориями пациентов повышена в 2,0-3,0 раза, инфаркта миокарда – в 2,4-3,3 раза, инсульта – в 2,0 раза, а сердечно-сосудистой смерти — в 1,14 раза [14-16]. Такие больные нуждаются в эффективной гиполипидемической терапии, но около трети из них самостоятельно отказываются от приема статинов в течение года после назначения [17]. Кроме того, больным СД 2 типа почти в 3 раза чаще требуется повторная реваскуляризация коронарных артерий, которая ассоциируется с более высоким риском тяжелых осложнений [18,19].
Принципиально новым подходом к лечению дислипидемии является применение моноклональных антител, блокирующих фермент PCSK9, который регулирует деградацию (разрушение) рецептора к ЛНП [20]. Одним из таких препаратов является алирокумаб [21]. В рандомизированных клинических исследованиях лечение алирокумабом в дополнение к статинам и эзетимибу или в качестве альтернативы другим гиполипидемическим средствам вызывало значительное снижение уровня ЛНП и позволяло достичь целевых значений липидов крови [22]. Приверженность к лечению в течение как минимум 71 недели была почти 100%. При этом отклонение от интервала введения (раз в две недели) на 3 дня не сказывалось на терапевтическом эффекте [23]. По данным мета-анализа, снижение риска сердечнососудистых событий при лечении ингибиторами PCSK9, добавленными к комбинированной терапии статинами и эзетимибом, составило 51%, а при стандартной терапии статинами и эзетимибом – только 6% [24]. Соответственно, при оценке технологий в здравоохранении с применением этих схем коэффициенты числа возможных событий в течение 2 лет после ОКС составили 0,94 при стандартной фармакотерапии и 0,49 при применении алирокумаба [25]. Два препарата этого класса – алирокумаб и эволокумаб – с 2018 г. включены в Перечень жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП) [26].
Существенное увеличение доступности препаратах этой группы в нашей стране возможно на условиях государственного (страхового) возмещения. Ранее проведенный анализ [25] показал экономическую целесообразность включения алирокумаба в Перечень ЖНВЛП. Целью данного исследования была оценка экономической целесообразности включения алирокумаба в Перечень лекарственных препаратов для льготных категорий граждан (Перечень ОНЛС) для лечения пациентов высокого сердечно-сосудистого риска.
Материал и методы
В качестве целевой группы выбраны пациенты с семейной гиперхолестеринемией, перенесшие ОКС и реваскуляризацию миокарда в течение последнего года, рефрактерные к применению статинов и эзетимиба в максимальных переносимых дозах и нуждающиеся в аферезе липидов [27]. Расчетные данные показывают, что такие больные составляют не менее 19,4% от общего числа больных, перенесших ОКС [9]. Отечественные данные об интенсификации липидснижающей терапии у пациентов высокого сердечнососудистого риска отсутствуют, поэтому для проведения расчетов использовали данные, полученные в США [27].
Горизонт анализа составил 2 года в соответствии с принятой методикой [28] и данными клинических исследований алирокумаба [27]. При определении прямых затрат учитывали режимы применения анализируемых препаратов, указанные в соответствующих исследованиях [29,30] и в официальных инструкциях по медицинскому применению (табл. 1) [31]. В качестве препарата сравнения при оценке затрат на стандартную гиполипидемическую терапию рассматривали аторвастатин в максимальной дозе 80 мг/сут (включенный в Перечень ОНЛС) [26] в комбинации с эзетимибом в дозе 10 мг в сутки (не включен в ЖНВЛП).
| Препарат | Режим применения | Стоимость упаковки с 10% НДС (руб.) |
|---|---|---|
| Алирокумаб (Пралуэнт) (раствор для подкожного введения 75 или 150 мг/мл №2) | 1 инъекция каждые 2 нед. | 29 260,81 |
| Эзетимиб (Эзетрол) (таблетки 10 мг № 28) | 10 мг/сут длительно | 1 784,94 |
| Аторвастатин (Липримар) (таблетки 80 мг №30 | 80 мг/сут длительно | 1 095,50 |
Средняя стоимость эзетимиба (Эзетрол) рассчитана с учетом оптовой цены за вычетом максимальной оптовой надбавки и с учетом НДС [32]. Стоимости алирокумаба (Пралуэнт) и аторвастатина (Липримар) определены на основании Регистра предельных отпускных цен с учетом НДС [33].
Анализ «влияние на бюджет». Анализ влияния на бюджет при включении алирокумаба в режимы фармакотерапии у пациентов целевой популяции проводили согласно утвержденным правилам [34] по сравнению с препаратами, уже включенными в Перечень ОНЛС. С использованием моделирования сравнивали две альтернативные медицинские технологии:
- текущий режим терапии с использованием статина в максимальных переносимых дозах и эзетемиба (целевых уровней ХС ЛНП достичь не удается);
- добавление к статинам и эзетимибу алирокумаба.
При недостаточной эффективности лечения показан аферез липидов плазмы крови, который проводится один раз в 2 недели. Модель Маркова, описывающая применение анализируемых технологий в рамках проводимого анализа, представлена на рис. 1.
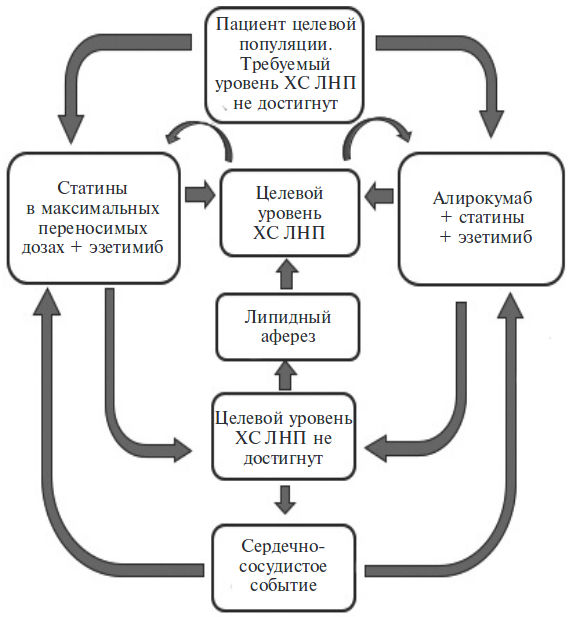
Для анализа влияния на бюджет учитывали прямые затраты на лекарственные препараты и аферез и прямые затраты на лечение сердечно-сосудистых событий.
В табл. 2. приведены данные о частоте применения афереза у пациентов, получающих алирокумаб и плацебо (т.е. только на фоне стандартной терапии) [35]. Согласно Программе государственных гарантий бесплатного оказания гражданам медицинской помощи на 2018 г. [36] стоимость одного дня лечения аферезом липидов плазмы крови в условиях дневного стационара за счет средств ОМС составляет 14 619,50 руб. Согласно КСГ №34 «Лечение наследственных атерогенных нарушений липидного обмена с применением афереза (липидная фильтрация, афинная и иммуносорбция липопротеидов) в случае отсутствия эффективности базисной терапии» предусмотрен коэффициент затрат 5,07, что составляет в сумме 74 120,86 руб./1 сеанс [37].
| Процент выполненных процедур афереза от необходимого числа | Средневзвешенный процент пациентов, получающих аферез, принятый для расчета | Частота афе реза в группе алиро кумаба, % | Частота афе реза в группе сравнения (%) |
|---|---|---|---|
| Аферез не проводится | 0 | 63,4 | 0 |
| >0% — ≤25% | 12,5 | 17,1 | 0 |
| >25% — ≤50% | 37,5 | 12,2 | 14,3 |
| >50% — ≤75% | 62,5 | 2,4 | 23,8 |
| >75% — ≤100 | 87,5 | 2,4 | 33,3 |
| 100% | 100 | 2,4 | 28,6 |
При оценке частоты сердечно-сосудистых событий учитывали базовую вероятность их наступления в группе, экстраполированную на целевую группу с глобальной модели по оценке влияния на бюджет при включении препарата алирокумаб в схему лечения (табл. 3).
| Событие | Вероятность наступления в год, % | Стоимость законченного случая (руб.) |
|---|---|---|
| Примечание: *Среднее между осложненной и неосложненной формой.**Средняя стоимость при равной частоте осложнений: тромбоэмболии легочной артерии, фибрилляции предсердий (с оперативным пособием и без), сердечно-легочная недостаточность | ||
| Нефатальный инфаркт миокарда | 9 | 52 520,14* |
| Нестабильная стенокардия | 1 | 80 374,94** |
| Реваскуляризация миокарда | 36 | 207 418,08 |
| Нефатальный инсульт | 2 | 120 297,40 |
| Сердечно-сосудистая смерть | 4 | — |
По данным мета-анализа М. Silverman и соавт. [38], снижение риска сердечно-сосудистых событий при использовании ингибиторов PCSK9 составляет 51%, а при лечении статином и эзетемибом – 6%. Эти показатели учитывали при расчете частоты сердечно-сосудистых событий в группах пациентов, получающих стандартную терапию и алирокумаб. Затраты на лечение сердечно-сосудистых событий рассчитывали, исходя из стоимости лечения одного законченного случая согласно тарифному соглашению ОМС по Москве 2017 г. (табл. 3) [39].
При расчете затрат на терапию учитывали смертность пациентов, в связи с которой они выбывали из исследования. Было сделано допущение, что число случаев смерти распределяется равномерно в течение года. Анализ чувствительности проводили для проверки устойчивости полученных результатов основного сценария к изменению входного параметра – стоимости препарата алирокумаб. Анализ «стоимости болезни» включал кроме вышеперечисленных затрат оценку непрямых расходов (недополучение валового внутреннего продукта – ВВП). Непрямые затраты в виде потери ВВП по причине смерти пациентов на период дожития до возраста завершения экономической активности (72 года) рассчитывали в соответствии с требованиями нормативных документов [40]. Расчет проводили для группы из 1000 пациентов с последующим перерасчетом на 1 пациента.
Размер недополученного ВВП рассчитывали путем деления годового ВВП страны на численность работоспособного населения и число рабочих дней в году. ВВП за 2016 г. составил 85 880,6 млрд руб, занятое население – 72 392 600 человек, число рабочих дней в году – 247. Для анализа были приняты следующие сроки временной нетрудоспособности при наступлении сердечно-сосудистых событий (согласно стандартам оказания медицинской помощи): нефатальный инфаркт миокарда – 80 дней, реваскуляризация миокарда – 30 дней, нефатальный инсульт – 80 дней [25]. Непрямые расходы, связанные с оплатой временной нетрудоспособности, получены умножением стоимости одного дня на соответствующее количество дней. Недополученный ВВП вследствие стойкой утраты трудоспособности или преждевременной смерти определен как отношение ВВП к трудоспособному населению. В 2016 г. он составил 488 782 руб. на человека трудоспособного возраста [41]. Эти показатели, как и соответствующий параметр при ОКС [42], отнесены к непрямым расходам.
Анализ минимизации затрат. Алирокумаб сравнивали с другим ингибитором PCSK9, зарегистрированным в России, – эволокумабом, применяющимся по сходным показаниям [43]. При отсутствии различий в эффекте должен применяться вариант анализа «затраты-эффект» – анализ минимизации затрат (CMA – Cost Minimization Analysis). Стоимость эволокумаба (Репата, Амджен Мэньюфэкчуринг Лимитед, Пуэрто-Рико, США), 140 мг/мл, шприцы 1 мл, рассчитана согласно методике определения предельной отпускной цены производителя [44], поскольку препарат включен в Перечень ЖНВЛП на 2018 г. (на момент анализа цена еще не была зарегистрирована). Минимальная цена определена в Словакии и составляет 386 евро за 2 шприца по 140 мг [45] или 579 евро в месяц (с учетом необходимости 3 шприцов), что по курсу ЦБ РФ на момент анализа (69,64 руб.) [46] соответствует 40 321,56 руб. С учетом таможенных сборов в 3,2% стоимость месяца лечения составляет 41 611,84 руб, а с учетом НДС (10%) – 45 773,03 руб.
Определяли показатель минимизации затрат CMR (Cost Minimization Ratio), показывающий, на сколько применение менее затратной медицинской технологии позволяет снизить затраты на лечение пациента [47]. Также оценивали показатель упущенных возможностей Q, характеризующий объем терапии, который мог бы быть обеспечен при использовании менее затратной медицинской технологии. Упомянутые показатели рассчитывали по формулам: CMR=DC2-DC1 и Q=CMR/DC1 × 100%,где CMR – показатель минимизации затрат; DC1 – прямые затраты при применении менее дорогой медицинской технологии; DC2 – прямые затраты при более дорогой медицинской технологии; Q – показатель упущенных возможностей.
Результаты
Определение целевой группы пациентов. В России ежегодно диагностируют в среднем 520 000 случаев ОКС [42]. Среди них расчетная доля больных с семейной гиперхолестеринемией составляет 19,4% (100 880 человек). ОКС с давностью 1 год и проведенной реваскуляризацией можно ожидать у 36% (на основании опроса врачей кардиологов), или 36 317 пациентов. Экстраполируя данные по частоте приема статинов пациентами с ОКС (88%) на эту группу пациентов, можно ее ограничить до 31 959 человек. Согласно данным модели интенсификации липидснижающей терапии у пациентов высокого сердечно-сосудистого риска [23], назначение алирокумаба потребуется примерно 14% из этих пациентов. Таким образом, размер целевой группы был ограничен 4 474 пациентами с семейной гиперхолестеринемией, перенесшими ОКС в течение последнего года и рефрактерными к терапии статинами. Частоту сердечнососудистых событий рассчитывали в целевой группе пациентов в рамках горизонта исследования 2 года. Результаты оценки прямых медицинских затрат на лечение пациентов целевой группы приведены в табл. 4.
| Показатели | Статин + эзетимиб | Алирокумаб + статин+эзетимиб |
|---|---|---|
| Первый год | ||
| Лекарственные затраты | 160,50 | 1 877,82 |
| Затраты на аферез | 6 463,67 | 1 848,94 |
| Затраты на лечение сердечнососудистых событий | 59,01 | 30,64 |
| Суммарно за первый год | 6 683,18 | 3 729,82 |
| Второй год | ||
| Лекарственные затраты | 154,52 | 1 812,93 |
| Затраты на аферез | 6 022,85 | 1 008,64 |
| Затраты на лечение сердечнососудистых событий | 57,00 | 30,15 |
| Суммарно за второй год | 6 234,37 | 2 851,72 |
| Суммарные затраты за 2 года | ||
| Прямые медицинские затраты | 12 917,55 | 6 581,54 |
| Разница (%) | — | Разница (%) |
Результаты анализа влияния на бюджет свидетельствуют о снижении суммарных прямых затрат на лечение пациентов целевой группы при добавлении алирокумаба к липидснижающей терапии на 48,6%. Это обусловлено преимущественно снижением частоты назначения дорогостоящей процедуры афереза при включении алирокумаба в схему лечения. Результаты анализа показали, что включение алирокумаба в Перечень ОНЛС экономически обосновано.
Анализ чувствительности. Наибольший вклад в суммарные прямые затраты при стандартном варианте терапии вносят затраты на липидный аферез, в то время как затраты на лекарственную терапию не превышают 2,5% от общей суммы. При добавлении к терапии алирокумаба затраты на его применение также начинают вносить значимый вклад в общие затраты. Как было отмечено выше, стоимость процедуры липидного афереза регламентируется тарифным соглашением по программе ОМС на 2018 г. В связи с этим единственным входным параметром, способным значимо повлиять на результат, является цена алирокумаба. Кроме того, выше было показано, что добавление алирокумаба в схему терапии позволяет снизить затраты. В связи с этим анализ чувствительности проводили для проверки устойчивости полученных результатов основного сце нария к однонаправленному изменению входного параметра: повышению стоимости алирокумаба. Полу ченный результат оказался устойчивым к возрастанию цены на алирокумаб (табл. 5). Предлагаемый режим терапии с применением алирокумаба остается менее затратным при увеличении цены на алирокумаб в 2,84 раза (на 184%).
| Увеличение цены алирокумаба | Разница затрат при использовании алирокумаба по сравнению с текущим вариантом терапии | |
|---|---|---|
| млн руб. | % | |
| 0% | -6 278,2 | -48,61 |
| +50% | -4 579,58 | -35,45 |
| +100% | -2 880,35 | -22,30 |
| +150% | -1 181,11 | -9,14 |
| +184% | -25,63 | -0,20 |
Анализ стоимости болезни. Учитывали прямые и непрямые затраты. Как указано выше, размер недополученного ВВП при невыходе пациента на работу в 2016 г. равнялся 4 802,90 руб. Средний возраст пациентов в клинических исследованиях составил 60-62 года (61 год) [23]. При расчетах было сделано допущение, что только 20% пациентов целевой группы относились к занятым в данной возрастной категории. При оценке недополученного ВВП за период дожития до 72 лет рассчитывали потери ВВП по причине смерти пациентов в течение первого и второго года исследования, а также за период дожития, т.е. в течение 9 лет. Включение алирокумаба в схемы терапии пациентов целевой группы позволит снизить бремя заболевания на 7,73 млрд руб, или на 56,1%, за 2 года терапии (табл. 6).
| Показатели | Статин + эзетимиб | Алирокумаб + статин + эзетимиб |
|---|---|---|
| Первый год | ||
| Прямые медицинские затраты | 6 683,18 | 3 729,82 |
| Недополученный ВВП, в связи с временной потерей трудоспособност | 39,50 | 20,52 |
| Недополученный ВВП в результате смерти в рамках горизонта исследования | 19,61 | 9,99 |
| Второй год | ||
| Прямые медицинские затраты | 6 234,37 | 6 234,37 |
| Недополученный ВВП, в связи с временной потерей трудоспособности | 38,07 | 20,19 |
| Недополученный ВВП в результате смерти в рамках горизонта исследования | 58,46 | 30,48 |
| Потери ВВП за годы дожития | ||
| Недополученный ВВП за годы дожития до возраста потери экономической активности (72 года) | 702,54 | 369,42 |
| Суммарные затраты за 2 года | ||
| Стоимость болезни | 13 775,73 | 6 050,53 |
| Разница (%) | — | -56,49 |
Анализ минимизации затрат. Исходя из обоснованного научного предположения об одинаковой эффективности (доля пациентов, достигающих целевых значений ХС ЛНП) алирокумаба и еще одного зарегистрированного в России препарата этого класса – эволокумаба – мы проводили анализ минимизации затрат. При этом стоимость статинов и эзетимиба не учитывали, поскольку считается, что дозировки и эффекты этих препаратов одинаковы в выделенных нами гипотетических группах алирокумаба и эволокумаба. Согласно официальным инструкциям у пациентов с семейной гиперхолестеринемией алирокумаб применяется в дозе 150-300 мг в месяц (2 инъекции), а эволокумаб – в дозе 280 мг (2 инъекции) или 420 мг в месяц (3 инъекции). Исходя из этого были рассчитаны лекарственные затраты на 52-недельную терапию сравниваемыми препаратами с последующим определением показателей минимизации затрат и упущенных возможностей. Результаты сравнения приведены в рис. 2. Расчетные данные показывают, что терапия алирокумабом по сравнению эволокумабом у пациентов рассматриваемой целевой группы позволит снизить затраты на 170 284 руб. на 1 пациента в год (-31,0%) или пролечить с тем же эффектом на 42,0% больше больных.
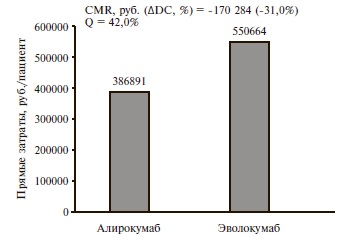
Обсуждение
При проведении анализа минимизации затрат сравнивали два ингибитора PCSK9, зарегистрированные в РФ: алирокумаб и эволокумаб. Этот вид анализа использовался потому, что нам не удалось обнаружить в доступной литературе результатов прямых сравнительных клинических исследований этих препаратов. Непрямое же сравнение не позволяет выявить достоверных различий между их клиническим эффектом, оцениваемым по доле пациентов, достигающих целевых уровней липидов крови. Опубликованный систематизированный обзор, показавший преимущество эволокумаба перед алирокумабом по степени снижения уровня холестерина ЛНП [48], не может быть использован для фармакоэкономического анализа, по нескольким причинам:
1. В мета-анализ не были включены некоторые исследования, в частности, RUTHERFORD и RUTHER
FORD-2 [49,50], что могло исказить результат непрямого сравнения в пользу эволокумаба. В группе плацебо не наблюдалось значительного повышения уровня холестерина ЛНП, поэтому включение исследований RUTHERFORD в сетевой мета-анализ могло повлиять на оценку общей эффективности эволокумаба по сравнению с плацебо.
2. В анализе не учтено изменение эффективности при титровании дозы алирокумаба.
3. В группах плацебо в исследованиях эволокумаба, включенных в мета-анализ, отмечалось выраженное повышение содержания холестерина ЛНП (почти на 13%) [51], в то время как в исследованиях алирокумаба уровни холестерина ЛНП в группах плацебо были стабильными или менялись очень незначительно [52,53].
4. Выбор временных интервалов для оценки эффективности не позволяет сравнить эффективность алирокумаба в этом анализе. Так, в программе ODYSSEY эффективность алирокумаба оценивали через 24 недели терапии [51]. Поскольку все данные по эффективности эволокумаба приведены для периода 10-12 недель, он теоретически получает дополнительные преимущества перед алирокумабом, так как для него не учитывается эффективность на большем временном горизонте.
5. В рассматриваемых исследованиях пациенты получали различную базовую терапию и значительно отличались по клинико-демографическим характеристикам, что не позволяет достоверно судить о сравнительной эффективности анализируемых препаратов.
6. В мета-анализе изменения уровня холестерина ЛНП сравнивали с контролем, а не с исходным уровнем, как это было сделано при оценке первичных конечных точке в анализируемых исследованиях.
Выводы
1. Включение алирокумаба в Перечень ОНЛС экономически обосновано. За 2 года применение алирокумаба должно снизить нагрузку на бюджет на 49,05% при лечении пациентов высокого сердечно-сосудистого риска, в том числе с семейной гиперхолестеринемией и рефрактерностью к гиполипидемической терапии по сравнению с лечением статинами и эзетимибом.
2. Полученный результат является устойчивым к росту цены на алирокумаб на 184% (в 2,89 раза).
3. Применение алирокумаба позволяет снизить бремя заболевания на 56,49%, или 7,78 млрд руб, в целевой группе за 2 года терапии по сравнению с текущей практикой лечения.
4. Курсовая стоимость лечения алирокумабом на 31,0% ниже таковой эволокумабом, что может позволить снизить затраты на лечение 1 пациента на 170 284 руб. в год и пролечить дополнительно 42% пациентов.
Используемые источники
- Bongard V, Dallongeville J, Arveiler D. et al. Attainment of low-density lipoprotein cholesterol target in the French general population according to levels of cardiovascular risk: insights from the MONA LISA study. Arch Cardiovasc Dis 2013;106(2):93–102.
- Bruckert E, Ferrières J. Evidence supporting primary prevention of cardiovascular diseases with statins: gaps between updated clinical results and actual practice. Arch Cardiovasc Dis 2014;107(3):188–200.
- Antiochos P, Marques-Vidal P, Waeber G, Vollenweider P. Five year trends in dyslipidaemia prevalence and management in Switzerland: the CoLaus study. Nutr Metab Cardiovasc Dis 2015;25(11):1007–15.
- Yan AT, Yan RT, Tan M, et al. Contemporary management of dyslipidemia in high-risk patients: targets still not met. Am J Med 2006;119(8):676–83.
- Joy TR, Hegele R. A. Narrative review: statin-related myopathy. Ann Intern Med 2009;150(12):858–68.
- Fernandez G, Spatz E. S, Jablecki C, Phillips PS. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med 2011;78(6):393–403.
- Nielsen SF, Nordestgaard BG. Negative statin-related news stories decrease statin persistence and increase myocardial infarction and cardiovascular mortality: a nationwide prospective cohort study. Eur Heart J 2016;37(11):908-16.
- Bittner V, Deng L, Rosenson RS, et al. Trends in the use of nonstatin lipid-lowering therapy among patients with coronary heart disease: a retrospective cohort study in the medicare population 2007 to 2011. J Am Coll Cardiol 2015;66(17): 1864–72.
- Gamboa CM, Safford MM, Levitan EB, et al. Statin underuse and low prevalence of LDL-C control among US adults at high risk of coronary heart disease. Am J Med Sci 2014;348(2):108–14.
- Толпыгина С.Н., Марцевич С.Ю. Изучение динамики частоты приема основных классов лекарственных препаратов, показанных при лечении пациентов с хронической ишемической болезнью сердца, с 2004 по 2014 г. данные регистра прогноз ИБС. Клиницист 2016;10(1): 29-35.
- Vonbank A, Agewall S, Kjeldsen KP, et al. Comprehensive efforts to increase adherence to statin therapy. Eur Heart . 2017 Jan 10. pii: ehw628.
- Krempf M, Simpson RJ, Ramey DR, et al. Patient and physician factors influence decision-making in hypercholesterolemia: a questionnaire-based survey. Lipids Health Dis 2015;14(1):45-7.
- Burggraaf B, Castro Cabezas M. Interventions in type 2 diabetes mellitus and cardiovascular mortality-An overview of clinical trials. Eur J Intern Med 2017;42:1-15.
- Дедов И.И., Александров А.A. Проблемы острого инфаркта миокарда у больных сахарным диабетом. Сахарный диабет 2008;1:4-10.
- Какорин С.В., Тулякова Э.В., Воронкова К.В., Мкртумян А.М. Острое нарушение мозгового кровообращения у больных сахарным диабетом 2 типа. Сахарный диабет 2013; (1):63–70.
- Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet 2010;375:2215–22.
- Simpson SH, Lin M, Eurich DT. Community pharmacy-based inducement programs associated with better medication adherence: a cohort study. Ann Pharmacother 2017;51(8):630-9.
- Ярбеков Р.Р., Чигогидзе Н.А., Сигаев И.Ю., Керен М.А. Ближайшая и отдаленная эффективность чрескожного коронарного вмешательства у больных ИБС с многососудистым поражением коронарных артерий и сахарным диабетом II типа. Анналы хирургии 2014; 5:21-26.
- Голухова Е.З., Кузнецова Е.В. Реваскуляризация миокарда у больных ИБС в сочетании с сахарным диабетом 2 типа: обзор современных технологий. Сахарный диабет 2016; 19(5):406-13.
- Wong ND, Rosenblit PD, Greenfield RS. Advances in dyslipidemia management for prevention of atherosclerosis: PCSK9 monoclonal antibody therapy and beyond. Cardiovasc Diagn Ther 2017;7 (Suppl 1):S11-20.
- Robinson JG, Farnier M, Krempf M. et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015;372(16):1489–99.
- El Shahawy M, Cannon CP, Blom DJ, et al. Efficacy and safety of Alirocumab versus ezetimibe over 2 years (from ODYSSEY COMBO II). Am J Cardiol 2017;17:31031-7.
- Farnier M, Colhoun HM, Sasiela WJ, et al. Long-term treatment adherence to the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab in 6 ODYSSEY Phase III clinical studies with treatment duration of 1 to 2 years. J Clin Lipidol 2017:S1933-2874.
- Silverman MG, Ference BA, Im K. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions. A systematic review and meta-analysis. JAMA 2016; 316(12):1289-97.
- Зырянов С.К., Дьяков И.Н. Клинико-экономическая экспертиза алирокумаба при рефрактерности к стандартной гиполипидемической терапии. Качественная клиническая практика 2016;4:4-13.
- Распоряжение Правительства Российской Федерации No2323-р от 23.10.2017.
- Farnier M, Gaudet D, Valcheva V, et al. Efficacy of alirocumab in high cardiovascular risk populations with or without heterozygous familial hypercholesterolemia: Pooled analysis of eight ODYSSEY Phase 3 clinical program trials. Intern J Cardiol 2016;223:750–7.
- Методические рекомендации по оценке влияния на бюджет в рамках реализации программы государственных гарантий бесплатного оказания гражданам медицинской помощи. М, ФГБУ “Центр экспертизы и контроля качества медицинской помощи”, 2016, 27С.
- Landmesser U, Chapman MJ, Farnier M, et al. on behalf of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). European Society of Cardiology/European Atherosclerosis Society Task Force consensus statement on proprotein convertase subtilisin/kexin type 9 inhibitors: practical guidance for use in patients at very high cardiovascular risk. Eur Heart J 2016;0:1–11. doi:10. 1093/eurheartj/ehw480.
- Nanchen D, Gencer B, Auer R. et al. Prevalence and management of familial hypercholesterolaemia in patients with acute coronary syndromes. Europ Heart J 2015;36:2438–45.
- Инструкция по медицинскому применению лекарственного препарата Пралуэнт.
- http://pharmindex. ru, дата обращения 13.12.2017.
- http://grls. rosminzdrav. ru, дата обращения к ресурсу 19.01.2018.
- Об утверждении Правил формирования перечней лекарственных препаратов для медицинского применения и минимального ассортимента лекарственных препаратов, необходимых для оказания медицинской помощи. Постановление Правительства РФ от 28.08.2014 N871.
- Moriarty PM, Parhofer KG, Babirak SP, et al. Alirocumab in patients with heterozygous familial hypercholesterolaemia undergoing lipoprotein apheresis: the ODYSSEY ESCAPE trial. Eur Heart J 2016;37(48):3588-95.
- О Программе государственных гарантий бесплатного оказания гражданам медицинской помощи на 2018 год и на плановый период 2019 и 2020 годов. Постановление Правительства РФ No1492 от 08.12.2017.
- Методические рекомендации по способам оплаты медицинской помощи за счет средств обязательного медицинского страхования. Минздрав России, Федеральный ФОМС. Утверждены на заседании рабочей группы МЗ РФ, протокол заседания No55/11/19 от 14.11.2017; http://www. ttfoms. tomsk. ru/document/ обращение к ресурсу 11.12.2017.
- Silverman MG, Ference BA, Im K, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA 2016;316(12):1289-97.
- Тарифное соглашение на оплату медицинской помощи, оказываемой по территориальной программе обязательного медицинского страхования города Москвы на 2017 год от 29 декабря 2016 г.
- Приказ Минэкономразвития России No192, Минздравсоцразвития России No323н, Минфина России No 45н, Росстата No113 от 10. 04. 2012 «Об утверждении методологии расчета экономических потерь от смертности, заболеваемости и инвалидизации населения».
- Дедов И.И., Концевая А.В., Шестакова М.В. и др. Экономические затраты на сахарный диабет 2 типа и его основные сердечно-сосудистые осложнения в Российской Федерации. Сахарный диабет 2016;19(6):518-27.
- Концевая А.В., Калинина А.М., Колтунов И.Е., Оганов Р.Г. Социальноэкономический ущерб от острого коронарного синдрома в Pоссийской Федерации. РФК 2011;7(2):158-66.
- Инструкция по медицинскому применению лекарственного препарата Репата.
- Правила государственной регистрации и перерегистрации устанавливаемых производителями лекарственных препаратов предельных отпускных цен на лекарственные препараты, включенные в Перечень ЖНВЛП. Утверждены Постановлением Правительства РФ No865 от 29.10.2010 в редакции Постановления No979 от 15.09.2015.
- http://www. health. gov. sk/Clanok?zuuc-201712-lieky; обращение к ресурсу 11.12.2017.
- http://www. cbr. ru/currency_base/daily. aspx?date_req=11.12.2017; обращение к ресурсу 11.12.2017.
- Фармакоэкономика и фармакоэпидемиология – практика примемлемых решений. Ред. В.Б.Герасимов, А.Л.Хохлов, О.И.Карпов. М.: Медицина, 2005, 352 с.
- Toth PP, Worthy G, Gandra SR, et al. Systematic review and network meta-analysis on the efficacy of evolocumab and other therapies for the management of lipid levels in hyperlipidemia. J Am Heart Assoc 2017;6(10).
- Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012;126:2408–17.
- Raal FJ, Stein EA, Dufour R, et al. RUTHERFORD-2 Investigators. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet 2015;385:331–40.
- Robinson JG, Nedergaard BS, Rogers WJ, et al; for the LAPLACE-2 Investigators. Effect of evolocumab or ezetimibe added to moderateor high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA 2014;311:1870–82.
- Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the PCSK9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J 2015;169: 906e915e13.
- Robinson JG, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015;372(16):148
Версия на английском языке
Фармакологическое действие
Полностью гуманизированное моноклональное антитело [изотип иммуноглобулинов G1 (IgG1)], мишенью которого является фермент пропротеиновая конвертаза субтилизин-кексин типа 9 (PCSK9). Алирокумаб производится с помощью технологии рекомбинантной ДНК с использованием суспензионной культуры клеток яичника китайского хомячка.
Алирокумаб имеет молекулярную массу приблизительно 146 кДа.
PCSK9 связывается с рецепторами ЛПНП (Р-ЛПНП) на поверхности гепатоцитов, способствуя деградации Р-ЛПНП в печени. Р-ЛПНП являются главными рецепторами, которые выводят из системного кровотока циркулирующие ЛПНП, поэтому уменьшение количества Р-ЛПНП при связывании их с PCSK9 приводит к повышению концентрации Хc-ЛПНП в крови. Ингибируя связывание PCSK9 с Р-ЛПНП, алирокумаб увеличивает количество Р-ЛПНП для выведения ЛПНП, снижая, таким образом, концентрации Хc-ЛПНП в крови.
Р-ЛПНП также связывают богатые триглицеридами (ТГ) ремнантные ЛПОНП и ЛППП. Поэтому лечение алирокумабом может снижать концентрации этих ремнантных липопротеинов, о чем свидетельствует их уменьшение в аполипопротеине В (Апо В), холестерине липопротеинов, не являющихся липопротеинами высокой плотности (Хc-ЛПнеВП) и ТГ. Алирокумаб также снижает концентрации липопротеинов а (ЛП(а)), являющихся формой ЛПНП, которые связаны с аполипопротеином (а). Однако было показано, что Р-ЛПНП имеют низкую аффинность к ЛП(а), в связи с чем точный механизм, с помощью которого алирокумаб снижает ЛП(а), полностью не установлен.
В генетических исследованиях, проведенных у человека, были выявлены разновидности гена PCSK9 с мутациями потери или повышения функции. У пациентов с одним аллелем PCSK9 с мутацией потери функции отмечались более низкие концентрации Хс-ЛПНП, которые коррелировали со значительно более низкой частотой развития ИБС. У некоторых пациентов были выявлены мутации потери функции в двух аллелях, и у них отмечались очень низкие концентрации Хс-ЛПНП в крови с концентрациями в крови Хс-ЛПВП и ТГ в нормальном диапазоне. Наоборот, мутации повышения функции в гене PCSK9 были выявлены у пациентов с повышенными концентрациями Хс-ЛПНП в крови и клиническим диагнозом семейной гиперхолестеринемии.
Наблюдательный анализ показал, что без лечения концентрации Хс-ЛПНП в крови у пациентов с мутациями повышения функции в гене PCSK9 находились в диапазоне, подобном наблюдавшемуся у пациентов с более часто встречающимися мутациями, вызывающими гетерозиготную форму семейной гиперхолестеринемии (такими как мутации в гене Р-ЛПНП), демонстрируя центральную роль фермента PCSK9 в метаболизме Хс-ЛПНП и его концентрациях в крови. В многоцентровом двойном слепом, плацебо-контролируемом исследовании продолжительностью 14 недель 13 пациентов с гетерозиготной формой семейной гиперхолестеринемией, связанной с мутацией повышения функции в гене PCSK9, были рандомизированы в 2 группы: группу, получающую алирокумаб в дозе 150 мг 1 раз в 2 недели, и группу, получающую плацебо. Среднее значение концентрации Хс-ЛПНП в крови составляло 151.5 мг/дл. На второй неделе лечения среднее значение снижения исходной концентрации Хс-ЛПНП в крови составило 62.5% в группе пациентов, получавших алирокумаб, по сравнению с 8.8% у пациентов, получавших плацебо. На 8-й неделе лечения среднее значение снижения концентрации Хс-ЛПНП в крови от исходного значения у всех пациентов, получавших алирокумаб, составило 72.4%.
Большое количество исследований, проведенных у человека и животных, продемонстрировали центральную роль, которую играют повышенные концентрации Хс-ЛПНП в крови в начале и при прогрессировании атеросклероза. Другие липопротеины, содержащие Апо В-100, особенно богатые триглицеридами ремнантные липопротеины (образовавшиеся из ЛПОНП и ЛППП) и ЛП(а), также считаются способствующими развитию атеросклероза. Однако проведенные до настоящего времени исследования не выявили независимого влияния снижения концентраций этих липопротеинов на сердечно-сосудистую заболеваемость и смертность.
В исследованиях in vitro алирокумаб не индуцировал антитело-зависимую клеточно-опосредованную токсичность и комплемент-зависимую цитотоксичность (Fc-опосредованную эффекторную функцию), как в присутствии, так и в отсутствии PCSK9. У алирокумаба, связанного с PCSK9, не наблюдалось образования нерастворимых иммунных комплексов, способных связывать протеины комплемента.
Фармакокинетика
После п/к введения в дозе 50-300 мг среднее время достижения Cmax алирокумаба в сыворотке крови составляло 3-7 дней. Фармакокинетика алирокумаба после однократного п/к введения в дозе 75 мг в область живота, бедра или плеча была подобной. По данным популяционного анализа фармакокинетических показателей абсолютная биодоступность алирокумаба после п/к введения составляла 85%.
Незначительно большее (2.1-2.7-кратное), чем пропорциональное дозе, увеличение концентраций алирокумаба, наблюдалось при двукратном увеличении дозы с 75 мг до 150 мг 1 раз в 2 недели.
Равновесное состояние достигалось после введения 2-3 доз с двукратным коэффициентом накопления.
После в/в введения Vd алирокумаба составлял 0.04-0.05 л/кг, что указывает на распределение алирокумаба, главным образом, в системе кровообращения.
Предполагается, что алирокумаб расщепляется на небольшие пептиды и отдельные аминокислоты.
У алирокумаба наблюдались 2 фазы выведения. В низких концентрациях элиминация происходит преимущественно через насыщаемую связь с мишенью (PCSK9), в то время как при более высоких концентрациях элиминация алирокумаба происходит преимущественно через ненасыщаемый протеолитический путь. По данным популяционного анализа фармакокинетических показателей средний Т1/2 алирокумаба составлял 17-20 дней у пациентов, получавших алирокумаб в монотерапии подкожно в дозах 75 мг 1 раз в 2 недели или 150 мг 1 раз в 2 недели. При одновременном применении со статинами средний Т1/2 алирокумаба составлял 12 дней.
Показания активного вещества
АЛИРОКУМАБ
Для длительного лечения взрослых пациентов с первичной гиперхолестеринемией (несемейной и гетерозиготной формой семейной гиперхолестеринемии) или смешанной дислипидемией, включая пациентов с сахарным диабетом 2 типа, в дополнение к диете, для снижения концентрации Хс-ЛПНП, общего Хс, Хс-ЛПнеВП, аполипопротеина В (Апо В), триглицеридов и ЛП(а) и повышения концентраций Хс-ЛПВП и Апо А-1.
Применяют в комбинации со статинами (ингибиторами ГМГ-КоА-редуктазы) в сочетании или без сочетания с другой липид-модифицирующей терапией при невозможности достижения у пациентов целевой концентрации Хс-ЛПНП при приеме максимально допустимой дозы статинов;
Применяют в качестве монотерапии или как дополнение к другой, не содержащей статинов липид-модифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению.
Режим дозирования
Вводят п/к в область бедра, живота или плеча.
Рекомендуется каждый раз менять места инъекций. Не следует вводить в область активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции. Не следует вводить препарат в то же место, в которое вводились другие лекарственные препараты.
Начальная доза составляет 75 мг 1 раз в 2 недели. У пациентов, которым требуется большее снижение концентрации Хс-ЛПНП (>60%), начальная доза препарата может составлять 150 мг 1 раз в 2 недели.
Дозу следует подбирать индивидуально на основании таких параметров как исходные значения Хс-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4 недели после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы.
Побочное действие
Со стороны иммунной системы: редко: — гиперчувствительность, аллергический васкулит.
Со стороны дыхательной системы: часто — субъективные симптомы и объективные признаки со стороны верхних дыхательных путей, включая боль в ротоглотке, ринорею, чихание.
Со стороны кожи и подкожных тканей: часто — кожный зуд; редко — крапивница, монетовидная экзема.
Прочие: часто — реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность.
Противопоказания к применению
Беременность, период грудного вскармливания; возраст до 18 лет ; повышенная чувствительность к алирокумабу.
Применение при беременности и кормлении грудью
Противопоказано применение при беременности и в период лактации (грудного вскармливания).
Применение у детей
Препарат противопоказан для применения у детей и подростков в возрасте до 18 лет
Применение у пожилых пациентов
Препарат разрешен для применения у пожилых пациентов
Особые указания
С осторожностью следует применять у пациентов с почечной недостаточностью тяжелой степени и при печеночной недостаточности тяжелой степени.
Лекарственное взаимодействие
В клинических исследованиях при применении алирокумаба в комбинации с аторвастатином или розувастатином не наблюдалось каких-либо значимых изменений концентраций статинов в крови при повторных введениях алирокумаба, что указывает на то, что алирокумаб не влияет на изоферменты цитохрома Р450 (главным образом, изоферменты CYP3A4 и CYP2C9) и белки-транспортеры, такие как Р-гликопротеин (P-gp) и ОАТР (белок транспортер органических анионов).
Статины и другая липид-модифицирующая терапия, как известно, повышают синтез PCSK9, белка, являющегося мишенью алирокумаба. Повышение концентрации PCSK9 может привести к уменьшению системной экспозиции алирокумаба. Однако это не влияет на продолжительность его действия при применении 1 раз в 2 недели.
Пралуэнт — инструкция по применению
Синонимы, аналоги
Статьи
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Регистрационный номер:
ЛП-004078 — 240317
Торговое наименование препарата:
Пралуэнт
Международное непатентованное наименование:
алирокумаб
Лекарственная форма:
раствор для подкожного введения.
Состав:
В одном шприце/шприц-ручке с дозировкой 75 мг/мл содержится:
активное вещество: алирокумаб — 75,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 1,241* мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
В одном шприце/шприц-ручке с дозировкой 150 мг/мл содержится:
активное вещество: алирокумаб — 150,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 0,931* мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
* — суммарное количество L-гистидина и L-гистидина гидрохлорида моногидрата в пересчете на L-гистидин.
Описание:
Прозрачная или слегка опалесцирующая, бесцветная или желтоватого цвета жидкость.
Фармакотерапевтическая группа:
полностью гуманизированное моноклональное антитело (IgGl). Ингибитор пропротеиновой конвертазы субтилизинкексин типа 9 (PCSK9)
Код АТХ:
С10АХ14
Фармакологические свойства
Фармакодинамика
Алирокумаб является полностью гуманизированным моноклональным согласовано антителом [изотип иммуноглобулинов G1 (IgGl)], мишенью которого является фермент пропротеиновая конвертаза субтилизин-кексин типа 9 (PCSK9). Алирокумаб производится с помощью технологии рекомбинантной ДНК с использованием суспензионной культуры клеток яичника китайского хомячка.
Алирокумаб имеет молекулярную массу приблизительно 146 кДа.
Механизм действия
PCSK9 связывается с рецепторами липопротеинов низкой плотности (Р-ЛПНП) на поверхности гепатоцитов, способствуя деградации Р-ЛПНП в печени. Р-ЛПНП являются главными рецепторами, которые выводят из системного кровотока циркулирующие ЛПНП, поэтому уменьшение количества Р-ЛПНП при связывании их с PCSK9 приводит к повышению концентрации холестерина ЛПНП (ХС-ЛПНП) в крови. Ингибируя связывание PCSK9 с Р-ЛПНП, алирокумаб увеличивает количество Р-ЛПНП для выведения ЛПНП, снижая, таким образом, концентрации ХС-ЛПНП в крови.
Р-ЛПНП также связывают богатые триглицеридами (ТГ) ремнантные липопротеины очень низкой плотности (ЛПОНП) и липопротеины промежуточной плотности (ЛППП). Поэтому лечение алирокумабом может снижать концентрации этих ремнантных липопротеинов, о чем свидетельствует их уменьшение в аполипопротеине В (Апо В), холестерине липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП) и ТГ. Алирокумаб также снижает концентрации липопротеинов а (Лп(а)), являющихся формой ЛПНП, которые связаны с аполипопротеином (а). Однако было показано, что Р-ЛПНП имеют низкую аффинность к Лп(а), в связи с чем точный механизм, с помощью которого алирокумаб снижает Лп(а), полностью не установлен.
Генетические исследования
В генетических исследованиях, проведенных у человека, были выявлены разновидности гена PCSK9 с мутациями потери или повышения функции. У пациентов с одним аллелем PCSK9 с мутацией потери функции отмечались более низкие концентрации ХС-ЛПНП, которые коррелировали со значительно более низкой частотой развития ишемической болезни сердца. У некоторых пациентов были выявлены мутации потери функции в двух аллелях, и у них отмечались очень низкие концентрации ХС-ЛПНП в крови с концентрациями в крови ХС-ЛПВП и ТГ в нормальном диапазоне. Наоборот, мутации повышения функции в гене PCSK9 были выявлены у пациентов с повышенными концентрациями ХС-ЛПНП в крови и клиническим диагнозом семейной гиперхолестеринемии.
Наблюдательный анализ показал, что без лечения концентрации ХС-ЛПНП в крови у пациентов с мутациями повышения функции в гене PCSK9 находились в диапазоне, подобном наблюдавшемуся у пациентов с более часто встречающимися мутациями, вызывающими гетерозиготную форму семейной гиперхолестеринемии (такими как мутации в гене Р-ЛПНП), демонстрируя центральную роль фермента PCSK9 в метаболизме ХС-ЛПНП и его концентрациях в крови. В многоцентровом двойном слепом, плацебо-контролируемом исследовании продолжительностью 14 недель 13 пациентов с гетерозиготной формой семейной гиперхолестеринемией, связанной с мутацией повышения функции в гене PCSK9, были рандомизированы в 2 группы: группу, получающую алирокумаб в дозе 150 мг 1 раз в 2 недели, и группу, получающую плацебо. Среднее значение концентрации ХС-ЛПНП в крови составляло 151,5 мг/дл. На второй неделе лечения среднее значение снижения исходной концентрации ХС-ЛПНП в крови составило 62,5 % в группе пациентов, получавших алирокумаб, по сравнению с 8,8 % у пациентов, получавших плацебо. На 8-й неделе лечения среднее значение снижения концентрации ХС-ЛПНП в крови от исходного значения у всех пациентов, получавших алирокумаб, составило 72,4 %.
Фармакодинамические свойства
Алирокумаб является полностью гуманизированным моноклональным антителом, которое подавляет активность PCSK9 как в исследованиях in vitro, так и в системах моделей in vivo. Большое количество исследований, проведенных у человека и животных, продемонстрировали центральную роль, которую играют повышенные концентрации ХС-ЛПНП в крови в начале и прогрессировании атеросклероза. Другие липопротеины, содержащие Апо В-100, особенно богатые триглицеридами ремнантные липопротеины (образовавшиеся из ЛПОНП и ЛППП) и Лп(а), также считаются способствующими развитию атеросклероза. Однако проведенные до настоящего времени исследования не выявили независимого влияния снижения концентраций этих липопротеинов на сердечно-сосудистую заболеваемость и смертность.
В исследованиях in vitro, алирокумаб не индуцировал антитело-зависимую клеточно-опосредованную токсичность и комплемент-зависимую цитотоксичность (Fc-опосредованную эффекторную функцию), как в присутствии, так и в отсутствии PCSK9. У алирокумаба, связанного с PCSK9, не наблюдалось образования нерастворимых иммунных комплексов, способных связывать протеины комплемента.
Клиническая эффективность/клинические исследования
Эффективность алирокумаба была изучена в 10 исследованиях III фазы (5 плацебо-контролируемых и 5 эзетимиб-контролируемых исследований), включавших 5296 рандомизированных пациентов с гиперхолестеринемией (несемейной и гетерозиготной формой семейной) или смешанной гиперхолестеринемией, из них 3188 пациентов были рандомизированы для приема алирокумаба. Три из этих 10 исследований были проведены исключительно у пациентов с гетерозиготной формой семейной гиперхолестеринемии. Большинство пациентов в программе клинических исследований III фазы принимали одновременно липид-модифицирующую терапию, состоящую из максимально переносимых доз статинов в сочетании или без сочетания с другими липид-модифицирующими видами лечения и 1 г, имели высокий и очень высокий сердечно-сосудистыи риск.
Два исследования были проведены у пациентов, которые не получали одновременно статины, включая одно исследование у пациентов с документированной непереносимостью статинов.
Два исследования (LONG TERM и HIGH FH), включающие в общей сложности 2416 пациентов, были проведены только с дозой 150 мг 1 раз в 2 недели. Восемь исследований были проведены с дозой 75 мг 1 раз в 2 недели и критерием для повышения дозы до 150 мг 1 раз в 2 недели на 12-й неделе было недостижение пациентами целевого значения концентрации ХС-ЛПНП в крови, основанного на степени их сердечно-сосудистого риска на 8-й неделе лечения.
Исходные демографические характеристики были хорошо сбалансированными между группами алирокумаба и контроля. Возраст пациентов во всех исследованиях находился в диапазоне от 18 до 89 лет (средний возраст составлял 60 лет); 38 % пациентов были женского пола, большинство пациентов были белой расы, 5 % были негроидной расы, 2 % были представителями азиатской расы; среднее значение индекса массы тела (ИМТ) составляло 30 кг/м2. В клинических исследованиях III фазы 31 % пациентов имели сахарный диабет 2 типа и 64 % пациентов имели в анамнезе ишемическую болезнь сердца.
Главной конечной точкой эффективности во всех клинических исследованиях III фазы было среднее значение снижения концентрации ХС-ЛПНП в крови на 24-й неделе по сравнению с плацебо и эзетимибом. Все исследования соответствовали их главной конечной точке.
В целом, применение алирокумаба приводило также к статистически значимо большему значению процента снижения концентраций ОХ, ХС-ЛПнеВП, Апо В и Лп(а) по сравнению с плацебо/эзетимибом, независимо от того получали или нет пациенты статины. Алирокумаб также снижал концентрации ТГ и увеличивал концентрации ХС-ЛПВП и Апо А-1 по сравнению с плацебо.
Снижение концентрации ХС-ЛПНП в крови наблюдалось во всех возрастных группах, у лиц обоего пола, при разных показателях индекса массы тела (ИМТ), у лиц разных рас, и с разными исходными концентрациями ХС-ЛПНП в крови. Результаты по эффективности были единообразными у пациентов с гетерозиготной формой семейной гиперхолестеринемии, без гетерозиготной семейной гиперхолестеринемии, у пациентов со смешанной дислипидемией и пациентов с сахарным диабетом. Снижение концентраций ХС-ЛПНП в крови было единообразным, независимо от того, принимались или не принимались пациентами одновременно статины, и от доз последних.
В группе алирокумаба по сравнению с группами плацебо или эзетимиба на 12-й и 24-й неделе статистически значимо у более высокого процента пациентов была достигнута концентрация ХС-ЛПНП в крови, составляющая <70 мг/дл.
В исследованиях с использованием схем титрования дозы, основанных на соответствующих критериях, большинство пациентов достигали целевого значения концентрации ХС-ЛПНП в крови (основанного на их степени сердечно-сосудистого риска) на дозе 75 мг 1 раз в 2 недели, и большинство пациентов продолжали лечение в дозе алирокумаба 75 мг 1 раз в 2 недели. Липидоснижающий эффект алирокумаба наблюдался в пределах 15 дней после применения первой дозы, достижение максимального эффекта наблюдалось приблизительно на 4 неделе. Эффективность сохранялась на протяжении всей продолжительности исследуемого лечения (до 78 недель в исследовании LONG TERM).
После прекращения введения алирокумаба не наблюдалось «синдрома отмены» и концентрация ХС-ЛПНП в крови постепенно возвращалась к исходным показателям.
Фармакокинетика
Абсорбция
После подкожного введения препарата Пралуэнт в дозе опт 50 мг до 300 мг среднее время достижения максимальной концентрации алирокумаба в сыворотке крови составляло 3-7 дней.
Фармакокинетика алирокумаба после однократного подкожного введения в дозе 75 мг в область живота, бедра или плеча была подобной. По данным популяционного анализа фармакокинетических показателей абсолютная биодоступность алирокумаба после подкожного введения составляла 85 %.
Незначительно большее (2,1-2,7-кратное), чем пропорциональное дозе, увеличение концентраций алирокумаба, наблюдалось при двукратном увеличении дозы с 75 мг до 150 мг 1 раз в 2 недели.
Равновесное состояние достигалось после введения 2-3 доз с двукратным коэффициентом накопления.
Распределение
После внутривенного введения объем распределения алирокумаба составлял 0,04-0,05 л/кг, что указывает на распределение алирокумаба, главным образом, в системе кровообращения.
Метаболизм
Специальные исследования метаболизма не проводились, так как алирокумаб является белком. Предполагается, что алирокумаб расщепляется на небольшие пептиды и отдельные аминокислоты.
Выведение
У алирокумаба наблюдались 2 фазы выведения. В низких концентрациях элиминация происходит преимущественно через насыщаемую связь с мишенью (PCSK9), в то время как при более высоких концентрациях элиминация алирокумаба происходит преимущественно через ненасыщаемый протеолитический путь. По данным популяционного анализа фармакокинетических показателей средний период полувыведения алирокумаба составлял 17-20 дней у пациентов, получавших алирокумаб в монотерапии подкожно в дозах 75 мг 1 раз в 2 недели или 150 мг 1 раз в 2 недели. При одновременном применении со статинами, средний период полувыведения алирокумаба составлял 12 дней.
Особые группы пациентов
Пол
По данным популяционного анализа фармакокинетических показателей половая принадлежность пациентов не влияла на фармакокинетику алирокумаба.
Пациенты пожилого возраста
По данным популяционного анализа фармакокинетических показателей, возраст ассоциировался с небольшим различием в системной экспозиции алирокумаба в равновесном состоянии без влияния на его эффективность и безопасность.
Масса тела
По данным популяционного анализа фармакокинетических показателей масса тела оказывала незначительное влияние на системную экспозицию алирокумаба без влияния на его эффективность и безопасность.
Пациенты детского возраста
Фармакокинетические эффекты алирокумаба у пациентов детского возраста к настоящему моменту не изучались.
Нарушения функции печени
В клиническом исследовании I фазы при однократном подкожном введении алирокумаба в дозе 75 мг фармакокинетические показатели алирокумаба у пациентов с печеночной недостаточностью легкой и средней степени тяжести были аналогичными таковым у пациентов с нормальной функцией печени. Доступные данные по фармакокинетике алирокумаба у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Нарушения функции почек
Так как нет сведений о том, что моноклональные антитела выводятся почками, не ожидается, что функциональное состояние почек повлияет на фармакокинетику алирокумаба. Популяционный анализ фармакокинетических показателей продемонстрировал, что нарушение функции почек легкой и средней степени тяжести не оказывало значимого воздействия на фармакокинетику алирокумаба. Данные по фармакокинетике алирокумаба у пациентов с тяжелыми нарушениями функции почек ограничены.
Расовая принадлежность
По данным популяционного анализа фармакокинетических показателей расовая принадлежность не оказывала влияния на фармакокинетику алирокумаба. После однократного подкожного введения алирокумаба в дозах 100-300 мг не наблюдалось значимых различий в системной экспозиции у здоровых добровольцев, являющихся японцами и представителями белой расы.
Показания к применению
Препарат Пралуэнт показан для длительного лечения взрослых пациентов с первичной гиперхолестеринемией (несемейной и гетерозиготной формой семейной гиперхолестеринемии) или смешанной дислипидемией, включая пациентов с сахарным диабетом 2 типа, в дополнение к диете, для снижения концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП), общего холестерина (общего-ХС), холестерина липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП), аполипопротеина В (Апо В), триглицеридов (ТГ) и липопротеина а (ЛПа) и повышения концентраций холестерина липопротеинов высокой плотности (ХС-ЛПВП) и аполипопротеина А-1 (Апо А-1). Препарат Пралуэнт показан:
- в комбинации со статинами (ингибиторами ГМГ-КоА-редуктазы) в сочетании или без сочетания с другой липид-модифицирующей терапией при невозможности достижения у пациентов целевой концентрации ХС-ЛПНП при приеме максимально допустимой дозы статинов;
- в монотерапии или как дополнение к другой, не содержащей статинов липид-модифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению.
Влияние препарата Пралуэнт на сердечно-сосудистую заболеваемость и смертность в настоящее время не установлено.
Противопоказания
- Повышенная чувствительность к алирокумабу или какому-либо вспомогательному веществу препарата.
- Беременность (эффективность и безопасность не установлены).
- Период грудного вскармливания (эффективность и безопасность не установлены).
- Детский возраст до 18 лет (эффективность и безопасность не установлены).
С осторожностью
- Почечная недостаточность тяжелой степени.
- Печеночная недостаточность тяжелой степени.
Применение при беременности и в период грудного вскармливания
Беременность
Отсутствуют данные по применению препарата Пралуэнт у беременных женщин.
Ожидается, что алирокумаб, как и другие антитела класса IgG, проникает через плацентарный барьер.
Во время беременности применение препарата Пралуэнт не рекомендуется.
Период грудного вскармливания
Неизвестно, проникает ли алирокумаб в грудное молоко у человека.
В связи с тем, что многие лекарственные препараты, и в том числе иммуноглобулины, проникают в грудное молоко у человека, применение препарата Пралуэнт у женщин в период грудного вскармливания не рекомендуется. При необходимости применения препарата Пралуэнт в данный период следует прекратить грудное вскармливание.
Способ применения и дозы
Начальная доза препарата Пралуэнт составляет 75 мг, которую вводят подкожно 1 раз в 2 недели. У пациентов, которым требуется большее снижение концентрации ХС-ЛПНП (> 60 %), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят подкожно 1 раз в 2 недели.
Дозу препарата Пралуэнт следует подбирать индивидуально на основании таких параметров как исходные значения ХС-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4 недели после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы.
В случае пропуска дозы пациент должен получить инъекцию, как можно скорее, и затем продолжить лечение через 2 недели со дня пропущенной дозы.
Особые группы пациентов
Дети
Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Пациенты пожилого возраста
У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. раздел «Особые указания)».
Печеночная недостаточность
У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность
У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела
Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде подкожных инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Побочное действие
Представленные ниже данные по безопасности отражают применение алирокумаба у 3340 пациентов, большинство из которых имели высокий или очень высокий риск развития сердечно-сосудистых заболеваний, и которые получали алирокумаб в дозе 75 мг или 150 мг в виде подкожных инъекций 1 раз в 2 недели с продолжительностью лечения до 18 месяцев (включая 2408 пациентов, получавших лечение алирокумабом в течение 52 недель, и 639 пациентов, получавших лечение алирокумабом в течение не менее 76 недель). Данные по безопасности основаны на объединенных результатах 9 плацебо-контролируемых исследований (4 исследования фазы II и 5 исследований фазы III (все исследования у пациентов, одновременно принимающих статины), и 5 контролируемых эзетимибом исследований фазы III (в 3-х из которых пациенты одновременно принимали статины).
Наиболее частыми нежелательными реакциями (>1 % пациентов, получавших препарат Пралуэнт) были реакции в месте введения препарата, субъективные симптомы и объективные признаки со стороны верхних дыхательных путей и кожный зуд.
Наиболее частыми нежелательными реакциями, приводящими к прекращению лечения у пациентов, получавших препарат Пралуэнт, были реакции в месте введения препарата.
Не наблюдалось различий в отношении профиля безопасности между двумя дозами (75 мг 1 раз в 2 недели и 150 мг 1 раз в 2 недели), использованными в программе клинических исследований фазы III.
В контролируемых исследованиях 1158 пациентов (34,7%), получавшие препарат Пралуэнт, были в возрасте >65 лет, и 241 пациент (7,2%), получавшие препарат Пралуэнт, были в возрасте >75 лет. Не наблюдалось достоверных различий по безопасности и эффективности препарата по мере увеличения возраста пациентов.
Нежелательные реакции о которых сообщалось при применении препарата Пралуэнт в объединенных контролируемых исследованиях
Нежелательные реакции представлены в соответствии с частотой, классификация которой рекомендована Всемирной Организацией Здравоохранения: очень часто (>1/10), часто (от > 1/100 до <1/10), нечасто (от >1/1 000 до <1/100), редко (от >1/10 000 до <1/1 000), очень редко (<1/10000), частота неизвестна (частота не может быть определена на основании имеющихся данных).
Нарушения со стороны иммунной системы
Редко: гиперчувствительность, аллергический васкулит.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения
Часто: субъективные симптомы и объективные признаки со стороны верхних дыхательных путей, включая боль в ротоглотке, ринорею, чихание.
Нарушения со стороны кожи и подкожных тканей
Часто: кожный зуд.
Редко: крапивница, монетовидная экзема.
Общие расстройства и нарушения в месте введения
Часто: реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность.
Описание отдельных нежелательных реакций
Реакции в месте введения препарата
Реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность, отмечались у 6,1 % пациентов, получавших лечение алирокумабом, по сравнению с 4,1 % в контрольной группе. Большинство реакций в месте введения препарата были преходящими и слабо выраженными. Частота прекращения лечения вследствие развития реакций в месте введения была сопоставимой в обеих группах (0,2 % в группе алирокумаба по сравнению с 0,3 % в контрольной группе).
Генерализованные аллергические реакции
Генерализованные аллергические реакции чаще отмечались в группе алирокумаба, чем в контрольной группе, главным образом, с различием в частоте развития кожного зуда. Кожный зуд был обычно слабо выраженным и преходящим. Кроме этого, в контролируемых клинических исследованиях сообщалось о развитии редких и иногда серьезных аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит.
Значения ХС-ЛПНП <25 мг/дл
В объединенных контролируемых исследованиях у 796 пациентов из 3340 пациентов (23,8 %), получавших лечение препаратом Пралуэнт, отмечались два последовательно полученных значения концентрации ХС-ЛПНП в крови < 25 мг/дл, включая 288 пациентов (8,6 %) с двумя последовательно полученными значениями концентрации ХС-ЛПНП <15 мг/дл.
Эти случаи, главным образом, наблюдались, когда пациенты начинали и продолжали лечение препаратом Пралуэнт в дозе 150 мг 1 раз в 2 недели, независимо от исходных значений концентрации ХС-ЛПНП в крови или реакции на лечение.
Не было выявлено нежелательных реакций, связанных с этими значениями концентрации ХС-ЛПНП в крови.
Сердечно-сосудистые осложнения
В настоящее время продолжается исследование сердечно-сосудистых исходов, в котором главной конечной точкой являются подтвержденные экспертизой большие нежелательные сердечно-сосудистые осложнения (БНССО), такие как коронарная смерть, инфаркт миокарда, ишемический инсульт и нестабильная стенокардия, потребовавшая госпитализации.
При запланированном анализе объединенных исследований фазы III у 110 пациентов (3,5 %) в группе алирокумаба и у 53 пациентов (3 %) в контрольной группе (плацебо или активный контроль) наблюдались следующие, подтвержденные экспертизой, сердечно-сосудистые осложнения, развившиеся во время лечения: смерть, связанная с ишемической болезнью сердца, инфаркт миокарда, ишемический инсульт, нестабильная стенокардия, потребовавшая госпитализации, госпитализация по поводу хронической сердечной недостаточности и реваскуляризация. Подтвержденные БНССО наблюдались у 52 из 3182 пациентов (1,6 %) в группе алирокумаба и у 33 из 1792 пациентов (1,8%) в контрольной группе (плацебо и активный контроль).
При запланированном окончательном анализе клинического исследования LONG TERM подтвержденные экспертизой сердечно-сосудистые осложнения, развившиеся во время лечения, наблюдались у 72 из 1550 пациентов (4,6%) в группе алирокумаба и у 43 из 788 пациентов (5,1 %) в группе плацебо; подтвержденные экспертизой БНССО наблюдались у 27 из 1550 пациентов (1,7 %) в группе алирокумаба и у 26 из 788 пациентов (3,3 %) в группе плацебо. Отношения рисков (ОР) рассчитывали ретроспективно; для всех сердечно-сосудистых осложнений отношения рисков = 0,91 (95% ДИ, 0,62-1,34); для БНССО отношения рисков = 0,52 (95% ДИ, 0,31-0,90).
Смертность от всех причин
В клинических исследованиях фазы III смертность от всех причин составляла 0,6% (20 из 3182 пациентов) в группе алирокумаба и 0,9% (17 из 1792 пациентов) в контрольной группе. Главной причиной смертельных исходов были сердечно-сосудистые осложнения.
Иммуногенность/антитела к алирокумабу
Как и все другие протеины, применяющиеся для лечения, алирокумаб обладает потенциальной иммуногенностью. В исследованиях фазы III у 4,8 % пациентов, получавших лечение алирокумабом, отмечалось образование антител к алирокумабу (АА), по сравнению с 0,6 % в контрольной группе (плацебо и эзетимиб). У большинства этих пациентов наблюдались преходящие реакции образования АА с низкими титрами без нейтрализующей активности. По сравнению с пациентами, которые были АА-негативными, пациенты с АА-позитивным статусом не продемонстрировали различий в системной экспозиции алирокумаба, эффективности или безопасности, за исключением более высокой частоты развития реакций в месте введения препарата. Только у 1,2% пациентов в группе алирокумаба были выявлены нейтрализующие антитела. Большинство этих пациентов имело только один положительный результат анализа на наличие нейтрализующих антител; 10 пациентов (0,3%) имели два или более положительных результата анализа на наличие нейтрализующих антител. Данные у этих пациентов не подтверждают корреляции между наличием нейтрализующих антител и эффективностью в отношении снижения концентрации ХС-ЛПНП в крови и безопасностью.
Данные по иммуногенности зависят от чувствительности и специфичности методики их определения, а также от других факторов. Кроме этого, на наблюдаемую частоту АА-позитивного результата анализа оказывают влияние несколько факторов, включая обработку забранных проб крови, время забора крови, одновременно принимаемые лекарственные препараты и основное заболевание. По этим причинам сравнение частоты возникновения АА с частотой возникновения антител к другим препаратам может быть некорректным.
Передозировка
В контролируемых клинических исследованиях не было выявлено никаких изменений безопасности при более частом введении доз, чем рекомендованный режим дозирования 1 раз в 2 недели.
Взаимодействие с другими лекарственными средствами
Влияние алирокумаба на другие лекарственные средства
Так как алирокумаб является биологическим веществом, не ожидается каких-либо фармакокинетических эффектов алирокумаба на другие лекарственные препараты.
В клинических исследованиях при применении алирокумаба в комбинации с аторвастатином или розувастатином не наблюдалось каких-либо значимых изменений концентраций статинов в крови при повторных введениях алирокумаба, что указывает на то, что алирокумаб не влияет на изоферменты цитохрома Р450 (главным образом, изоферменты CYP3A4 и CYP2C9) и белки-транспортеры, такие как Р-гликопротеин (P-gp) и ОАТР (белок транспортер органических анионов).
Влияние других лекарственных средств на алирокумаб
Статины и другая липид-модифицирующая терапия, как известно, повышают синтез PCSK9, белка, являющегося мишенью алирокумаба. Повышение концентрации PCSK9 может привести к уменьшению системной экспозиции алирокумаба. Однако это не влияет на продолжительность действия препарата при применении алирокумаба 1 раз в 2 недели.
Особые указания
Аллергические реакции
В клинических исследованиях сообщалось о развитии генерализованных аллергических реакциях, включая зуд; также имелись сообщения о редких и иногда серьезных случаях аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит. При появлении симптомов и признаков серьезных аллергических реакций лечение препаратом Пралуэнт должно быть прекращено и следует начать проведение соответствующей симптоматической терапии.
Влияние на фертильность
Данные о неблагоприятном воздействии алирокумаба на фертильность отсутствуют.
Пожилые пациенты
Данные о применении алирокумаба у пациентов старше 75 лет ограничены. В контролируемых исследованиях 1158 пациентов (34,7%), получавших препарат Пралуэнт, были в возрасте >65 лет, и 241 пациент (7,2 %), получавших препарат Пралуэнт, были в возрасте >75 лет. Значимых различий в безопасности и эффективности препарата Пралуэнт с увеличением возраста не наблюдалось.
Почечная недостаточность
В клинических исследованиях количество пациентов с почечной недостаточностью тяжелой степени (клиренс креатинина <30 мл/мин/1,73 м2) было ограничено. Препарат Пралуэнт следует применять с осторожностью у пациентов с почечной недостаточностью тяжелой степени (см. раздел «С осторожностью»).
Печеночная недостаточность
Исследований алирокумаба у пациентов с печеночной недостаточностью тяжелой степени (класс С по шкале Чайлда-Пью) не проводились. Препарат Пралуэнт следует применять с осторожностью у данной категории пациентов (см. раздел «С осторожностью»).
Влияние на способность управлять транспортными средствами и механизмами
Препарат Пралуэнт не влияет или почти не влияет на способность управлять транспортными средствами и работать с механизмами.
Форма выпуска
Раствор для подкожного введения 75 мг/мл и 150 мг/мл.
По 1 мл в одноразовый шприц из прозрачного стекла (тип I), снабженный несъемной иглой из нержавеющей стали, защищенной колпачком из мягкого полимера.
По 1 шприцу в пластиковый блистер с покрытием.
По 1 блистеру или по 2 соединенных блистера или по 3 пары соединенных блистеров с инструкцией по применению в картонную пачку.
По 1 шприцу в шприц-ручку. По 1, 2 или 6 шприц-ручек с инструкцией по применению в картонную пачку, снабженную фиксатором.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Условия хранения
Хранить при температуре от 2°С до 8°С. Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Не применять по истечении срока годности, указанного на упаковке.
Условия отпуска
Отпускают по рецепту.
Производитель
Санофи Винтроп Индустрия, Франция (шприцы).
Юридическое лино, на имя которого выдано регистрационное удостоверение
АО Санофи-авентис груп, Франция.
Фасовщик (первичная упаковка)
Санофи Винтроп Индустрия, Франция (шприцы).
Упаковщик (вторичная (потребительская) упаковка) Санофи Винтроп Индустрия, Франция (шприцы).
Санофи-Авентис Дойчланд ГмбХ, Германия (шприц-ручки).
ЗАО «Санофи-Авентис Восток», Россия (шприц-ручки)
Выпускающий контроль качества
Санофи Винтроп Индустрия, Франция (шприцы).
Sanofi Winthrop Industrie, France.
1051 boulevard Industriel 76580 Le Trait, France.
Санофи-Авентис Дойчланд ГмбХ, Германия (шприц-ручки).
Sanofi-Aventis Deutschland GmbH, Germany.
Brueningstrasse 50/Industriepark Hoechst H500, H590, H600 65926 Frankfort am Main, Germany.
ЗАО «Санофи-Авентис Восток», Россия (шприц-ручки).
302516, Россия, Орловская область, Орловский район, с/п Болынекуликовское, ул. Ливенская, д. 1.
Претензии потребителей направлять по адресу в России:
Представительство АО «Санофи-авентис труп». 125009, г. Москва, ул. Тверская, 22.
В случае упаковки препарата на ЗАО «Санофи-Авентис Восток», Россия претензии потребителей направлять по адресу:
302516, Россия, Орловская область, Орловский район, с/п Болынекуликовское, ул. Ливенская, д. 1.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
П/к. Начальная доза препарата Пралуэнт составляет 75 мг, которую вводят 1 раз в 2 нед или 300 мг 1 раз каждые 4 нед (ежемесячно). У пациентов, которым требуется большее снижение концентрации Хс-ЛПНП (>60%), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят 1 раз в 2 нед.
Дозу препарата Пралуэнт следует подбирать индивидуально, на основании таких параметров, как исходные значения Хс-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4–8 нед после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы. При необходимости дополнительного снижения концентрации Хс-ЛПНП у пациентов, которым препарат Пралуэнт назначался в дозе 75 мг 1 раз каждые 2 нед или 300 мг 1 раз каждые 4 нед, доза может быть скорректирована до максимальной дозы 150 мг 1 раз каждые 2 нед.
В случае пропуска дозы пациент должен получить инъекцию как можно скорее и затем продолжить лечение по исходной схеме.
Особые группы пациентов
Дети. Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Пожилой возраст. У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. «Особые указания»).
Печеночная недостаточность. У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется.
Данные по применению препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность. У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела. Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде п/к инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
При назначении дозы 300 мг препарат вводят в виде двух инъекций по 150 мг в разные места.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Пралуэнт, 75 мг, раствор для п/к введения в предварительно заполненной шприц-ручке.
Пралуэнт, 150 мг, раствор для п/к введения в предварительно заполненной шприц-ручке.
Инструкция по применению
Пациента необходимо информировать о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору или позвонить по номеру телефона Представительства АО «Санофи-авентис груп», указанному в инструкции.
Части шприц-ручки указаны на рисунке

Важная информация
— шприц-ручка предназначена только для одноразового использования;
— шприц-ручка с зеленой кнопкой: в 1 мл раствора содержится 75 мг алирокумаба;
— шприц-ручка с серой кнопкой: в 1 мл раствора содержится 150 мг алирокумаба;
— препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
— шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
— шприц-ручку следует хранить в недоступном для детей месте;
— перед использованием шприц-ручки следует внимательно прочитать инструкцию;
— необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручки;
— неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
— трогать желтый защитный колпачок;
— использовать шприц-ручку при протекании или повреждении;
— использовать шприц-ручку при отсутствии голубого колпачка или если он ненадежно закреплен;
— использовать шприц-ручку повторно;
— трясти шприц-ручку;
— замораживать шприц-ручку;
— подвергать шприц-ручку воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед введением препарата пациенту понадобятся:
— шприц-ручка с препаратом Пралуэнт;
— салфетки, смоченные спиртом;
— ватные тампоны или марля;
— контейнер, резистентный к проколам.
1. Проверить этикетку на шприц-ручке:
— убедиться, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц-ручка с зеленой кнопкой — для дозы 75 мг/мл и шприц-ручка с серой кнопкой — для дозы 150 мг/мл);

— проверить срок годности. Не использовать по истечении срока годности.
2. Проверить окно:
— проверить, чтобы видимая в окно жидкость была прозрачной и бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А);
— пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата;
— не использовать шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок B).

3. Оставить шприц-ручку согреваться при комнатной температуре в течение 30–40 мин:
— не нагревать шприц-ручку, она должна согреться сама;
— использовать шприц-ручку как можно скорее после того, как она нагрелась;
— не класть шприц-ручку обратно в холодильник.
4. Подготовка места введения препарата:
— вымыть руки водой с мылом и вытереть их полотенцем;
— можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);

— пациент может стоять или сидеть во время введения препарата;
— продезинфицировать место введения салфеткой, смоченной спиртом;
— не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
— не вводить препарат в места рядом с видимыми венами;
— использовать разные места при каждом введении препарата.
— не вводить препарат в одно и то же место с другими препаратами.
Этап B. Как вводить препарат
1. После завершения всех пунктов «Этап А. Подготовка к введению» снять голубой колпачок:
— не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
— не надевать голубой колпачок обратно.

2. Держать шприц-ручку, как показано на рисунке:
— не трогать желтый защитный колпачок;
— убедиться, что видно окно.

3. Прижать желтый защитный колпачок к коже примерно под углом 90°:
— прижимать шприц-ручку к коже и удерживать ее так, чтобы желтый защитный колпачок стал невидимым (вдавленным в кожу). Шприц-ручка не будет работать, если желтый защитный колпачок не вдавлен в кожу полностью;

— при необходимости следует зажать кожу, чтобы убедиться, что место введения плотное.
4. Нажать большим пальцем на зеленую (если используется шприц-ручка Пралуэнт, 75 мг) или серую кнопку (если используется шприц-ручка Пралуэнт, 150 мг) и сразу отпустить ее:
— будет слышен щелчок. Введение препарата началось;
— окно начнет желтеть.

5. Продолжать удерживать шприц-ручку плотно прижатой к коже после того, как отпущена кнопка:
— введение препарата может продолжаться до 20 с.

6. Проверить, что окно стало желтого цвета до того, как убрана шприц-ручка с кожи:
— не убирать шприц-ручку с кожи до тех пор, пока все окно не будет желтого цвета;
— введение препарата завершено, после того как все окно будет желтого цвета и будет слышен второй щелчок;
— если окно не стало полностью желтого цвета, не следует вводить самостоятельно вторую дозу без консультации врача.

7. Убрать шприц-ручку с кожи:
— не растирать кожу после введения препарата;
— если пациент видит кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.

8. Утилизировать шприц-ручку и колпачок:
— не надевать голубой колпачок повторно;
— поместить шприц-ручку в контейнер, резистентный к проколам;

— всегда хранить контейнер в недоступном для детей месте.
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Пралуэнт, 75 мг, раствор для п/к введения в предварительно заполненном шприце.
Пралуэнт, 150 мг, раствор для п/к введения в предварительно заполненном шприце.
Части шприца показаны на рисунке.
Важная информация
Шприц предназначен для одноразового использования.

— шприц с зеленым поршнем: в 1 мл раствора содержится 75 мг алирокумаба;
— шприц с серым поршнем: в 1 мл раствора содержится 150 мг алирокумаба;
— препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
— шприц можно использовать только для однократной инъекции, и он должен быть утилизирован после использования.
Правила использования
— шприц следует хранить в недоступном для детей месте;
— перед использованием шприца следует внимательно прочитать инструкцию;
— необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприца;
— неиспользованные шприцы следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
— трогать иглу;
— использовать шприц при протекании или повреждении;
— использовать шприц при отсутствии серого колпачка для иглы, или если он ненадежно закреплен;
— использовать шприц повторно;
— трясти шприц;
— замораживать шприц;
— подвергать шприц воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед началом введения пациенту понадобятся:
— шприц с препаратом Пралуэнт;
— салфетки, смоченные спиртом;
— ватные тампоны или марля;
— контейнер, резистентный к проколам.
1. Перед началом следует:
— взять шприц из пакета, удерживая его за корпус.

2. Проверить этикетку на шприце:
— проверить, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц с зеленым поршнем — для дозы 75 мг/мл и шприц с серым поршнем — для дозы 150 мг/мл);
— проверить срок годности, не использовать по истечении срока годности;
— проверить, чтобы жидкость в шприце была прозрачной и бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц;
— проверить, что шприц не открыт и не поврежден.
3. Оставить шприц согреваться при комнатной температуре в течение 30–40 мин:
— не нагревать шприц, он должен согреться сам;
— использовать шприц как можно скорее после того, как он согреется;
— не класть шприц обратно в холодильник.
4. Подготовка места введения:
— вымыть руки водой и мылом и вытереть их полотенцем;
— можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);

— пациент может стоять или сидеть во время введения препарата;
— продезинфицировать место введения салфеткой, смоченной спиртом;
— не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
— не вводить препарат в места рядом с видимыми венами;
— использовать разные места при каждом введении препарата.
Этап B. Как вводить препарат
1. После завершения всех пунктов «Этап А. Подготовка к введению» снять колпачок для иглы:

— не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
— держать шприц за середину корпуса так, чтобы игла была направлена от пациента;
— не прикасаться к поршню;
— пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата. Не следует избавляться ни от каких пузырьков в шприце до начала инъекции;
— не надевать серый колпачок обратно.
2. При необходимости зажать кожу:
— большим и указательным пальцем зажать кожу так, чтобы получилась складка кожи в месте введения;

— удерживать кожную складку во время проведения всей инъекции.
3. Ввести иглу в складку кожи быстрым движением:

— использовать угол в 90° при сжатии 5 см кожи; использовать угол в 45° при сжатии 2 см кожи.
4. Опустить поршень вниз:
— ввести весь раствор медленным и равномерным нажатием на поршень;

5. Перед тем как удалить иглу, следует убедиться, что шприц пуст:
— не удалять шприц, пока он полностью не опустеет;
— вытаскивать иглу из кожи следует под тем же углом, под каким она была введена;
— не растирать кожу после введения препарата;
— если видна кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.

6. Утилизировать шприц и колпачок:
— не надевать серый колпачок повторно;
— не использовать шприц повторно;
— поместить шприц в контейнер, резистентный к проколам;
— всегда хранить контейнер в недоступном для детей месте.

Подготовка и обращение с препаратом
— пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления им медицинскими работниками информации по правильной технике проведения п/к инъекций;
— препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать;
— перед использованием предварительно заполненных шприц-ручек и предварительно заполненных шприцев следует согреть их до комнатной температуры;
— препарат Пралуэнт следует использовать как можно скорее, после того как он согреется. Препарат может храниться не более 24 ч при температуре не выше 25 °C;
— после использования шприц-ручки и шприцы следует поместить в резистентный к проколам контейнер и утилизировать согласно местным (государственным) требованиям;
— не использовать контейнер повторно;
— всегда следует хранить контейнер в местах, недоступных для детей;
— пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприц-ручки и предварительно заполненные шприцы;
— пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата;
— пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприц-ручек или шприцев, и они должны быть проинструктированы, как безопасно утилизировать их после использования.
Praluent
Регистрационный номер
Торговое наименование
Пралуэнт
Международное непатентованное наименование
Лекарственная форма
раствор для подкожного введения
Состав
В одном шприце/шприц-ручке с дозировкой 75 мг/мл содержится:
действующее вещество: алирокумаб — 75,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 1,241* мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
В одном шприце /шприц-ручке с дозировкой 150 мг/мл содержится:
действующее вещество: алирокумаб — 150,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 0,931 * мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
* — суммарное количество L-гистидина и L-гистидина гидрохлорида моногидрата в пересчёте на L-гистидин.
Описание
Прозрачная или слегка опалесцирующая, бесцветная или желтоватого цвета жидкость.
Фармакотерапевтическая группа
Код АТХ
Фармакологические свойства
Фармакодинамика
Алирокумаб является полностью гуманизированным моноклональным антителом [изотип иммуноглобулинов G1 (IgG1)], мишенью которого является фермент пропротеиновая конвертаза субтилизин-кексин типа 9 (PCSK9). Алирокумаб производится с помощью технологии рекомбинантной ДНК с использованием суспензионной культуры клеток яичника китайского хомячка.
Алирокумаб имеет молекулярную массу приблизительно 146 кДа.
Механизм действия
PCSK9 связывается с рецепторами липопротеинов низкой плотности (Р- ЛПНП) на поверхности гепатоцитов, способствуя деградации Р-ЛПНП в печени. Р-ЛПНП являются главными рецепторами, которые выводят из системного кровотока циркулирующие ЛПНП, поэтому уменьшение количества Р-ЛПНП при связывании их с PCSK9 приводит к повышению концентрации холестерина ЛПНП (ХС-ЛПНП) в крови. Ингибируя связывание PCSK9 с Р-ЛПНП, алирокумаб увеличивает количество Р-ЛПНП для выведения ЛПНП, снижая, таким образом, концентрации ХС-ЛПНП в крови.
Р-ЛПНП также связывают богатые триглицеридами (ТГ) ремнантные липопротеины очень низкой плотности (ЛПОНП) и липопротеины промежуточной плотности (ЛППП). Поэтому лечение алирокумабом может снижать концентрации этих ремнантных липопротеинов, о чем свидетельствует их уменьшение в аполипопротеине В (Апо В), холестерине липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП) и ТГ. Алирокумаб также снижает концентрации липопротеинов а (Лп(а)), являющихся формой ЛПНП, которые связаны с аполипопротеином (а). Однако было показано, что Р-ЛПНП имеют низкую аффинность к Лп(а), в связи с чем точный механизм, с помощью которого алирокумаб снижает Лп(а), полностью не установлен.
Генетические исследования
В генетических исследованиях, проведённых у человека, были выявлены разновидности гена PCSK9 с мутациями потери или повышения функции. У пациентов с одним аллелем PCSK9 с мутацией потери функции отмечались более низкие концентрации ХС-ЛПНП, которые коррелировали со значительно более низкой частотой развития ишемической болезни сердца. У некоторых пациентов были выявлены мутации потери функции в двух аллелях, и у них отмечались очень низкие концентрации ХС-ЛПНП в крови с концентрациями в крови ХС-ЛПВП и ТГ в нормальном диапазоне. Наоборот, мутации повышения функции в гене PCSK9 были выявлены у пациентов с повышенными концентрациями ХС-ЛПНП в крови и клиническим диагнозом семейной гиперхолестеринемии.
Наблюдательный анализ показал, что без лечения концентрации ХС-ЛПНП в крови у пациентов с мутациями повышения функции в гене PCSK9 находились в диапазоне, подобном наблюдавшемуся у пациентов с более часто встречающимися мутациями, вызывающими гетерозиготную форму семейной гиперхолестеринемии (такими как мутации в гене Р-ЛПНП), демонстрируя центральную роль фермента PCSK9 в метаболизме ХС-ЛПНП и его концентрациях в крови. В многоцентровом двойном слепом, плацебо-контролируемом исследовании продолжительностью 14 недель 13 пациентов с гетерозиготной формой семейной гиперхолестеринемией, связанной с мутацией повышения функции в гене PCSK9, были рандомизированы в 2 группы: группу, получающую алирокумаб в дозе 150 мг 1 раз в 2 недели, и группу, получающую плацебо. Среднее значение концентрации ХС-ЛПНП в крови составляло 151,5 мг/дл. На второй неделе лечения среднее значение снижения исходной концентрации ХС-ЛПНП в крови составило 62,5 % в группе пациентов, получавших алирокумаб, по сравнению с 8,8 % у пациентов, получавших плацебо. На 8-й неделе лечения среднее значение снижения концентрации ХС-ЛПНП в крови от исходного значения у всех пациентов, получавших алирокумаб, составило 72,4 %.
Фармакодинамические свойства
Алирокумаб является полностью гуманизированным моноклональным антителом, которое подавляет активность PCSK9 как в исследованиях in vitro, так и в системах моделей in vivo. Большое количество исследований, проведённых у человека и животных, продемонстрировали центральную роль, которую играют повышенные концентрации ХС-ЛПНП в крови в начале и прогрессировании атеросклероза. Другие липопротеины, содержащие Апо В-100, особенно богатые триглицеридами ремнантные липопротеины (образовавшиеся из ЛПОНП и ЛППП) и Лп(а), также считаются способствующими развитию атеросклероза. Однако проведённые до настоящего времени исследования не выявили независимого влияния снижения концентраций этих липопротеинов на сердечно-сосудистую заболеваемость и смертность.
В исследованиях in vitro, алирокумаб не индуцировал антитело-зависимую клеточно-опосредованную токсичность и комплемент-зависимую цитотоксичность (Fc-опосредованную эффекторную функцию), как в присутствии, так и в отсутствии PCSK9. У алирокумаба, связанного с PCSK9, не наблюдалось образования нерастворимых иммунных комплексов, способных связывать протеины комплемента.
Клиническая эффективность/клинические исследования
Эффективность алирокумаба была изучена в 10 исследованиях III фазы (5 плацебо-контролируемых и 5 эзетимиб-контролируемых исследований), включавших 5296 рандомизированных пациентов с гиперхолестеринемией (несемейной и гетерозиготной формой семейной) или смешанной гиперхолестеринемией, из них 3188 пациентов были рандомизированы для приёма алирокумаба. Три из этих 10 исследований были проведены исключительно у пациентов с гетерозиготной формой семейной гиперхолестеринемии. Большинство пациентов в программе клинических исследований III фазы принимали одновременно липид-модифицирующую терапию, состоящую из максимально переносимых доз статинов в сочетании
или без сочетания с другими липид-модифицирующими видами лечения и имели высокий и очень высокий сердечно-сосудистый риск. Два исследования были проведены у пациентов, которые не получали одновременно статины, включая одно исследование у пациентов с документированной непереносимостью статинов.
Два исследования (LONG TERM и HIGH FH), включающие в общей сложности 2416 пациентов, были проведены только с дозой 150 мг 1 раз в 2 недели. Восемь исследований были проведены с дозой 75 мг 1 раз в 2 недели и критерием для повышения дозы до 150 мг 1 раз в 2 недели на 12-й неделе было недостижение пациентами целевого значения концентрации ХС-ЛПНП в крови, основанного на степени их сердечно-сосудистого риска на 8-й неделе лечения.
Исходные демографические характеристики были хорошо сбалансированными между группами алирокумаба и контроля. Возраст пациентов во всех исследованиях находился в диапазоне от 18 до 89 лет (средний возраст составлял 60 лет); 38 % пациентов были женского пола, большинство пациентов были белой расы, 5 % были негроидной расы, 2 % были представителями азиатской расы; среднее значение индекса массы тела (ИМТ) составляло 30 кг/м2. В клинических исследованиях III фазы 31 % пациентов имели сахарный диабет 2 типа и 64 % пациентов имели в анамнезе ишемическую болезнь сердца.
Главной конечной точкой эффективности во всех клинических исследованиях III фазы было среднее значение снижения концентрации ХС-ЛПНП в крови на 24-й неделе по сравнению с плацебо и эзетимибом. Все исследования соответствовали их главной конечной точке.
В целом, применение алирокумаба приводило также к статистически значимо большему значению процента снижения концентраций ОХ, ХС-ЛПнеВП, Апо В и Лп(а) по сравнению с плацебо/эзетимибом, независимо от того получали или нет пациенты статины. Алирокумаб также снижал концентрации ТГ и увеличивал концентрации ХС-ЛПВП и Апр А-1 по сравнению с плацебо.
Снижение концентрации ХС-ЛПНП в крови наблюдалось во всех возрастных группах, у лиц обоего пола, при разных показателях индекса массы тела (ИМТ), у лиц разных рас, и с разными исходными концентрациями ХС- ЛПНП в крови. Результаты по эффективности были единообразными у пациентов с гетерозиготной формой семейной гиперхолестеринемии, без гетерозиготной семейной гиперхолестеринемии, у пациентов со смешанной дислипидемией и пациентов с сахарным диабетом. Снижение концентраций ХС-ЛПНП в крови было единообразным, независимо от того, принимались или не принимались пациентами одновременно статины, и от доз последних.
В группе алирокумаба по сравнению с группами плацебо или эзетимиба на 12-й и 24-й неделе статистически значимо у более высокого процента пациентов была достигнута концентрация ХС-ЛПНП в крови, составляющая <70 мг/дл.
В исследованиях с использованием схем титрования дозы, основанных на соответствующих критериях, большинство пациентов достигали целевого значения концентрации ХС-ЛПНП в крови (основанного на их степени сердечно-сосудистого риска) на дозе 75 мг 1 раз в 2 недели, и большинство пациентов продолжали лечение в дозе алирокумаба 75 мг 1 раз в 2 недели. Липидоснижающий эффект алирокумаба наблюдался в пределах 15 дней после применения первой дозы, достижение максимального эффекта наблюдалось приблизительно на 4 неделе. Эффективность сохранялась на протяжении всей продолжительности исследуемого лечения (до 78 недель в исследовании LONG TERM).
После прекращения введения алирокумаба не наблюдалось «синдрома отмены» и концентрация ХС-ЛПНП в крови постепенно возвращалась к исходным показателям.
Фармакокинетика
Абсорбция
После подкожного введения препарата Пралуэнт в дозе от 50 мг до 300 мг среднее время достижения максимальной концентрации алирокумаба в сыворотке крови составляло 3–7 дней.
Фармакокинетика алирокумаба после однократного подкожного введения в дозе 75 мг в область живота, бедра или плеча была подобной.
По данным популяционного анализа фармакокинетических показателей абсолютная биодоступность алирокумаба после подкожного введения составляла 85 %.
Незначительно большее (2,1-2,7-кратное), чем пропорциональное дозе, увеличение концентраций алирокумаба, наблюдалось при двукратном увеличении дозы с 75 мг до 150 мг 1 раз в 2 недели.
Равновесное состояние достигалось после введения 2-3 доз с двукратным коэффициентом накопления.
Распределение
После внутривенного введения объём распределения алирокумаба составлял 0,04-0,05 л/кг, что указывает на распределение алирокумаба, главным образом, в системе кровообращения.
Метаболизм
Специальные исследования метаболизма не проводились, так как алирокумаб является белком. Предполагается, что алирокумаб расщепляется на небольшие пептиды и отдельные аминокислоты.
Выведение
У алирокумаба наблюдались 2 фазы выведения. В низких концентрациях элиминация происходит преимущественно через насыщаемую связь с мишенью (PCSK9), в то время как при более высоких концентрациях элиминация алирокумаба происходит преимущественно через ненасыщаемый протеолитический путь. По данным популяционного анализа фармакокинетических показателей средний период полувыведения алирокумаба составлял 17-20 дней у пациентов, получавших алирокумаб в монотерапии подкожно в дозах 75 мг 1 раз в 2 недели или 150 мг 1 раз в 2 недели. При одновременном применении со статинами, средний период полувыведения алирокумаба составлял 12 дней.
Особые группы пациентов
Пол
По данным популяционного анализа фармакокинетических показателей половая принадлежность пациентов не влияла на фармакокинетику алирокумаба.
Пациенты пожилого возраста
По данным популяционного анализа фармакокинетических показателей, возраст ассоциировался с небольшим различием в системной экспозиции алирокумаба в равновесном состоянии без влияния на его эффективность и безопасность.
Масса тела
По данным популяционного анализа фармакокинетических показателей масса тела оказывала незначительное влияние на системную экспозицию алирокумаба без влияния на его эффективность и безопасность.
Пациенты детского возраста
Фармакокинетические эффекты алирокумаба у пациентов детского возраста к настоящему моменту не изучались.
Нарушения функции печени
В клиническом исследовании I фазы при однократном подкожном введении алирокумаба в дозе 75 мг фармакокинетические показатели алирокумаба у пациентов с печёночной недостаточностью лёгкой и средней степени тяжести были аналогичными таковым у пациентов с нормальной функцией печени. Доступные данные по фармакокинетике алирокумаба у пациентов с печёночной недостаточностью тяжёлой степени отсутствуют.
Нарушения функции почек
Так как нет сведений о том, что моноклональные антитела выводятся почками, не ожидается, что функциональное состояние почек повлияет на фармакокинетику алирокумаба. Популяционный анализ фармакокинетических показателей продемонстрировал, что нарушение функции почек лёгкой и средней степени тяжести не оказывало значимого воздействия на фармакокинетику алирокумаба. Данные, по фармакокинетике алирокумаба у пациентов с тяжёлыми нарушениями функции почек ограничены.
Расовая принадлежность
По данным популяционного анализа фармакокинетических показателей расовая принадлежность не оказывала влияния на фармакокинетику алирокумаба. После однократного подкожного введения алирокумаба в дозах 100-300 мг не наблюдалось значимых различий в системной экспозиции у здоровых добровольцев, являющихся японцами и представителями белой расы.
Показания
Препарат Пралуэнт показан для длительного лечения взрослых пациентов с первичной гиперхолестеринемией (несемейной и гетерозиготной формой семейной гиперхолестеринемии) или смешанной дислипидемией, включая пациентов с сахарным диабетом 2 типа, в дополнение к диете, для снижения концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП), общего холестерина (общего-ХС), холестерина липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП), аполипопротеина В (Апо В), триглицеридов (ТГ) и липопротеина а (ЛПа) и повышения концентраций холестерина липопротеинов высокой плотности (ХС-ЛПВП) и аполипопротеина А-1 (Апо А-1).
Препарат Пралуэнт показан:
— в комбинации со статинами (ингибиторами ГМГ-КоА-редуктазы) в сочетании или без сочетания с другой липид-модифицирующей терапией при невозможности достижения у пациентов целевой концентрации ХС-ЛПНП при приёме максимально допустимой дозы статинов;
— в монотерапии или как дополнение к другой, не содержащей статинов липид-модифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению.
Влияние препарата Пралуэнт на сердечно-сосудистую заболеваемость и смертность в настоящее время не установлено.
Противопоказания
- Повышенная чувствительность к алирокумабу или какому-либо вспомогательному веществу препарата.
- Беременность (эффективность и безопасность не установлены).
- Период грудного вскармливания (эффективность и безопасность не установлены).
- Детский возраст до 18 лет (эффективность и безопасность не установлены).
С осторожностью
- Почечная недостаточность тяжёлой степени.
- Печёночная недостаточность тяжёлой степени.
Применение при беременности и в период грудного вскармливания
Беременность
Отсутствуют данные по применению препарата Пралуэнт у беременных женщин.
Ожидается, что алирокумаб, как и другие антитела класса IgG, проникает через плацентарный барьер.
Во время беременности применение препарата Пралуэнт не рекомендуется.
Период грудного вскармливания
Неизвестно, проникает ли алирокумаб в грудное молоко у человека.
В связи с тем, что многие лекарственные препараты, и в том числе иммуноглобулины, проникают в грудное молоко у человека, применение препарата Пралуэнт у женщин в период грудного вскармливания не рекомендуется. При необходимости применения препарата Пралуэнт в данный период следует прекратить грудное вскармливание.
Способ применения и дозы
Начальная доза препарата Пралуэнт составляет 75 мг, которую вводят подкожно 1 раз в 2 недели. У пациентов, которым требуется большее снижение концентрации ХС-ЛПНП (> 60 %), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят подкожно 1 раз в 2 недели.
Дозу препарата Пралуэнт следует подбирать индивидуально на основании таких параметров как исходные значения ХС-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4 недели после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы.
В случае пропуска дозы пациент должен получить инъекцию, как можно скорее, и затем продолжить лечение через 2 недели со дня пропущенной дозы.
Особые группы пациентов
Дети
Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Пациенты пожилого возраста
У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. раздел «Особые указания)».
Печёночная недостаточность
У пациентов с печёночной недостаточностью лёгкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата Пралуэнт у пациентов с печёночной недостаточностью тяжёлой степени отсутствуют.
Почечная недостаточность
У пациентов с почечной недостаточностью лёгкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата у пациентов с почечной недостаточностью тяжёлой степени ограничены.
Масса тела
Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде подкожных инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Побочное действие
Представленные ниже данные по безопасности отражают применение алирокумаба у 3340 пациентов, большинство из которых имели высокий или очень высокий риск развития сердечно-сосудистых заболеваний, и которые получали алирокумаб в дозе 75 мг или 150 мг в виде подкожных инъекций 1 раз в 2 недели с продолжительностью лечения до 18 месяцев (включая 2408 пациентов, получавших лечение алирокумабом в течение 52 недель, и 639 пациентов, получавших лечение алирокумабом в течение не менее 76 недель). Данные по безопасности основаны на объединенных результатах 9 плацебо-контролируемых исследований (4 исследования фазы II и 5 исследований фазы III (все исследования у пациентов, одновременно принимающих статины), и 5 контролируемых эзетимибом исследований фазы III (в 3-х из которых пациенты одновременно принимали статины).
Наиболее частыми нежелательными реакциями (≥ 1 % пациентов, получавших препарат Пралуэнт) были реакции в месте введения препарата, субъективные симптомы и объективные признаки со стороны верхних дыхательных путей и кожный зуд.
Наиболее частыми нежелательными реакциями, приводящими к прекращению лечения у пациентов, получавших препарат Пралуэнт, были реакции в месте введения препарата.
Не наблюдалось различий в отношении профиля безопасности между двумя дозами (75 мг 1 раз в 2 недели и 150 мг 1 раз в 2 недели), использованными в программе клинических исследований фазы III.
В контролируемых исследованиях 1158 пациентов (34,7 %), получавшие препарат Пралуэнт, были в возрасте ≥ 65 лет, и 241 пациент (7,2 %), получавшие препарат Пралуэнт, были в возрасте ≥75 лет. Не наблюдалось достоверных различий по безопасности и эффективности препарата по мере увеличения возраста пациентов.
Нежелательные реакции, о которых сообщалось при применении препарата Пралуэнт в объединенных контролируемых исследованиях
Нежелательные реакции представлены в соответствии с частотой, классификация которой рекомендована Всемирной Организацией Здравоохранения: очень часто (≥ 1/10), часто (от ≥ 1/100 до < 1/10), нечасто (от ≥ 1/1 000 до < 1/100), редко (от ≥1/10 000 до <1/1 000), очень редко (< 1/10000), частота неизвестна (частота не может быть определена на основании имеющихся данных).
Нарушения со стороны иммунной системы
Редко: гиперчувствительность, аллергический васкулит.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения
Часто: субъективные симптомы и объективные признаки со стороны верхних дыхательных путей, включая боль в ротоглотке, ринорею, чихание.
Нарушения со стороны кожи и подкожных тканей
Часто: кожный зуд.
Редко: крапивница, монетовидная экзема.
Общие расстройства и нарушения в месте введения
Часто: реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отёк, боль/болезненную чувствительность.
Описание отдельных нежелательных реакций
Реакции в месте введения препарата
Реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отёк, боль/болезненную чувствительность, отмечались у 6,1 % пациентов, получавших лечение алирокумабом, по сравнению с 4,1 % в контрольной группе. Большинство реакций в месте введения препарата были преходящими и слабо выраженными. Частота прекращения лечения вследствие развития реакций в месте введения была сопоставимой в обеих группах (0,2 % в группе алирокумаба по сравнению с 0,3 % в контрольной группе).
Генерализованные аллергические реакции
Генерализованные аллергические реакции чаще отмечались в группе алирокумаба, чем в контрольной группе, главным образом, с различием в частоте развития кожного зуда. Кожный зуд был обычно слабо выраженным и преходящим. Кроме этого, в контролируемых клинических исследованиях сообщалось о развитии редких и иногда серьёзных аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит.
Значения ХС-ЛПНП < 25 мг/дл
В объединенных контролируемых исследованиях у 796 пациентов из 3340 пациентов (23,8 %), получавших лечение препаратом Пралуэнт, отмечались два последовательно полученных значения концентрации ХС-ЛПНП в крови < 25 мг/дл, включая 288 пациентов (8,6 %) с двумя последовательно полученными значениями концентрации ХС-ЛПНП < 15 мг/дл.
Эти случаи, главным образом, наблюдались, когда пациенты начинали и продолжали лечение препаратом Пралуэнт в дозе 150 мг 1 раз в 2 недели, независимо от исходных значений концентрации ХС-ЛПНП в крови или реакции на лечение.
Не было выявлено нежелательных реакций, связанных с этими значениями концентрации ХС-ЛПНП в крови.
Сердечно-сосудистые осложнения
В настоящее время продолжается исследование сердечно-сосудистых исходов, в котором главной конечной точкой являются подтвержденные экспертизой большие нежелательные сердечно-сосудистые осложнения (БНССО), такие как коронарная смерть, инфаркт миокарда, ишемический инсульт и нестабильная стенокардия, потребовавшая госпитализации.
При запланированном анализе объединенных исследований фазы III у 110 пациентов (3,5 %) в группе алирокумаба и у 53 пациентов (3 %) в контрольной группе (плацебо или активный контроль) наблюдались следующие, подтвержденные экспертизой, сердечно-сосудистые осложнения, развившиеся во время лечения: смерть, связанная с ишемической болезнью сердца, инфаркт миокарда, ишемический инсульт, нестабильная стенокардия, потребовавшая госпитализации, госпитализация по поводу хронической сердечной недостаточности и реваскуляризация. Подтвержденные БНССО наблюдались у 52 из 3182 пациентов (1,6 %) в группе алирокумаба и у 33 из 1792 пациентов (1,8 %) в контрольной группе (плацебо и активный контроль).
При запланированном окончательном анализе клинического исследования LONG TERM подтвержденные экспертизой сердечно-сосудистые осложнения, развившиеся во время лечения, наблюдались у 72 из 1550 пациентов (4,6 %) в группе алирокумаба и у 43 из 788 пациентов (5,1 %) в группе плацебо; подтвержденные экспертизой БНССО наблюдались у 27 из 1550 пациентов (1,7 %) в группе алирокумаба и у 26 из 788 пациентов (3,3 %) в группе плацебо. Отношения рисков (ОР) рассчитывали ретроспективно; для всех сердечно-сосудистых осложнений отношения рисков = 0,91 (95 % ДИ, 0,62-1,34); для БНССО отношения рисков = 0,52 (95 % ДИ, 0,31-0,90).
Смертность от всех причин
В клинических исследованиях фазы III смертность от всех причин составляла 0,6 % (20 из 3182 пациентов) в группе алирокумаба и 0,9 % (17 из 1792 пациентов) в контрольной группе. Главной причиной смертельных исходов были сердечно-сосудистые осложнения.
Иммуногенность/антитела к алирокумабу
Как и все другие протеины, применяющиеся для лечения, алирокумаб обладает потенциальной иммуногенностью. В исследованиях фазы III у 4,8 % пациентов, получавших лечение алирокумабом, отмечалось образование антител к алирокумабу (АА), по сравнению с 0,6 % в контрольной группе (плацебо и эзетимиб). У большинства этих пациентов наблюдались преходящие реакции образования АА с низкими титрами без нейтрализующей активности. По сравнению с пациентами, которые были АА-негативными, пациенты с АА-позитивным статусом не продемонстрировали различий в системной экспозиции алирокумаба, эффективности или безопасности, за исключением более высокой частоты развития реакций в месте введения препарата. Только у 1,2 % пациентов в группе алирокумаба были выявлены нейтрализующие антитела. Большинство этих пациентов имело только один положительный результат анализа на наличие нейтрализующих антител; 10 пациентов (0,3 %) имели два или более положительных результата анализа на наличие нейтрализующих антител. Данные у этих пациентов не подтверждают корреляции между наличием нейтрализующих антител и эффективностью в отношении снижения концентрации ХС-ЛПНП в крови и безопасностью.
Данные по иммуногенности зависят от чувствительности и специфичности методики их определения, а также от других факторов. Кроме этого, на наблюдаемую частоту АА-позитивного результата анализа оказывают влияние несколько факторов, включая обработку забранных проб крови, время забора крови, одновременно принимаемые лекарственные препараты и основное заболевание. По этим причинам сравнение частоты возникновения АА с частотой возникновения антител к другим препаратам может быть некорректным.
Передозировка
В контролируемых клинических исследованиях не было выявлено никаких изменений безопасности при более частом введении доз, чем рекомендованный режим дозирования 1 раз в 2 недели.
Взаимодействие с другими лекарственными средствами
Влияние алирокумаба на другие лекарственные средства
Так как алирокумаб является биологическим веществом, не ожидается каких- либо фармакокинетических эффектов алирокумаба на другие лекарственные препараты.
В клинических исследованиях при применении алирокумаба в комбинации с аторвастатином или розувастатином не наблюдалось каких-либо значимых изменений концентраций статинов в крови при повторных введениях алирокумаба, что указывает на то, что алирокумаб не влияет на изоферменты цитохрома P450 (главным образом, изоферменты CYP3A4 и CYP2C9) и белки-транспортёры, такие как Р-гликопротеин (P-gp) и ОАТР (белок транспортёр органических анионов).
Влияние других лекарственных средств на алирокумаб
Статины и другая липид-модифицирующая терапия, как известно, повышают синтез PCSK9, белка, являющегося мишенью алирокумаба. Повышение концентрации PCSK9 может привести к уменьшению системной экспозиции алирокумаба. Однако это не влияет на продолжительность действия препарата при применении алирокумаба 1 раз в 2 недели.
Особые указания
Аллергические реакции
В клинических исследованиях сообщалось о развитии генерализованных аллергических реакциях, включая зуд; также имелись сообщения о редких и иногда серьёзных случаях аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит. При появлении симптомов и признаков серьёзных аллергических реакций лечение препаратом Пралуэнт должно быть прекращено и следует начать проведение соответствующей симптоматической терапии.
Влияние на фертильность
Данные о неблагоприятном воздействии алирокумаба на фертильность отсутствуют.
Пожилые пациенты
Данные о применении алирокумаба у пациентов старше 75 лет ограничены. В контролируемых исследованиях 1158 пациентов (34,7 %), получавших препарат Пралуэнт, были в возрасте ≥ 65 лет, и 241 пациент (7,2 %), получавших препарат Пралуэнт, были в возрасте ≥ 75 лет. Значимых различий в безопасности и эффективности препарата Пралуэнт с увеличением возраста не наблюдалось.
Почечная недостаточность
В клинических исследованиях количество пациентов с почечной недостаточностью тяжёлой степени (клиренс креатинина <30 мл/мин/1,73 м2) было ограничено. Препарат Пралуэнт следует применять с осторожностью у пациентов с почечной недостаточностью тяжёлой степени (см. раздел «С осторожностью»).
Печёночная недостаточность
Исследований алирокумаба у пациентов с печёночной недостаточностью тяжёлой степени (класс C по шкале Чайлда-Пью) не проводились. Препарат Пралуэнт следует применять с осторожностью у данной категории пациентов (см. раздел «С осторожностью»).
Влияние на способность управлять транспортными средствами, механизмами
Препарат Пралуэнт не влияет или почти не влияет на способность управлять транспортными средствами и работать с механизмами.
Форма выпуска
Раствор для подкожного введения 75 мг/мл и 150 мг/мл.
По 1 мл в одноразовый шприц из прозрачного стекла (тип I), снабжённый несъемной иглой из нержавеющей стали, защищённой колпачком из мягкого полимера.
По 1 шприцу в пластиковый блистер с покрытием.
По 1 блистеру или по 2 соединенных блистера или по 3 пары соединенных блистеров с инструкцией по применению в картонную пачку.
По 1 шприцу в шприц-ручку. По 1, 2 или 6 шприц-ручек с инструкцией по применению в картонную пачку, снабжённую фиксатором.
Хранение
Хранить при температуре от 2 °C до 8 °C. Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Не применять по истечении срока годности, указанного на упаковке.
Условия отпуска из аптек
Отпускают по рецепту.