Rec.INN
зарегистрированное ВОЗ
Лекарственное взаимодействие
Входит в состав препаратов:
список
Фармакологическое действие
Иммунодепрессант, ингибитор интерлейкина. Представляет собой рекомбинантную версию антагониста рецептора интерлейкина-1 (IL-1ra) человеческого, продуцированного штаммом E.coli с использованием технологии рекомбинантной ДНК.
Блокирует биологическую активность интелейкина-1α (ИЛ-1α) и интерлейкина-1β (ИЛ-1β) путем конкурентного ингибирования их связей с рецептором интерлейкина-1 типа I. Интерлейкин-1 (ИЛ-1) является ключевым провоспалительным цитокином, медиатором многих клеточных ответов, включая те, которые имеют важное значение при синовиальном воспалении.
Анакинра ингибирует реакции, индуцированные ИЛ-1 in vitro, в т.ч. индукцию оксида азота и простагландина Е2 и/или синтез коллагеназы синовиальными клетками, фибробластами и хондроцитами.
Спонтанные мутации в гене CIAS1/NLRP3 были обнаружены у большинства пациентов с криопирин-ассоциированными периодическими синдромами (CAPS). CIAS1/NLRP3 кодирует криопирин, компонент инфламмасомы. Активация инфламмасомы приводит к протеолитической матурации и секреции ИЛ-1β, который вызывает широкий спектр реакций, включая системное воспаление. У пациентов с CAPS, не получавших лечение, отмечают повышенный уровень С-реактивного белка (СРБ), амилоида А (САА) и ИЛ-6 в сыворотке крови по сравнению с нормальными уровнями. Было отмечено, что применение анакинры приводит к снижению уровней острофазных белков и уменьшению экспрессии ИЛ-6. Снижение уровней острофазных белков было зарегистрировано в течение первых недель лечения.
Фармакокинетика
После п/к введения анакинры в дозе 70 мг абсолютная биодоступность у здоровых добровольцев (n = 11) составляет 95%. Процесс абсорбции является фактором, ограничивающим скорость выведения анакинры из плазмы после п/к инъекции.
У пациентов на фоне лечения Cmax анакинры в плазме достигалась через 3-7 ч после п/к введения в клинически значимых дозах (от 1 до 2 мг/кг; n=18). Концентрация в плазме уменьшалась без очевидной фазы распределения, а T1/2 составлял от 4 до 6 ч. У пациентов, получавших анакинру кумуляция не наблюдалась после ежедневного п/к введения в течение до 24 недель. Средние (SD) показатели клиренса и Vd по результатам популяционного фармакокинетического анализа данных 35 пациентов составили 105 (27) мл/мин и 18.5 (11) л соответственно. Выводится почками. Клиренс анакинры возрастает с увеличением КК.
Показания активного вещества
АНАКИНРА
Взрослым, подросткам и детям в возрасте от 8 месяцев и старше с массой тела от 10 кг и выше: лечение криопирин-ассоциированных периодических синдромов (CAPS), включая мультисистемное воспалительное заболевание неонатального возраста (NOMID)/хронический младенческий неврологический кожно-артикулярный синдром (CINCA); синдром Макла-Уэллса (MWS); семейный холодовой аутовоспалительный синдром (FCAS).
Режим дозирования
Для п/к введения.
Лечение проводит врач-специалист, имеющий опыт диагностики и лечения CAPS.
Для взрослых, подростков и детей в возрасте от 8 месяцев и старше с массой тела от 10 кг и выше начальная доза составляет 1-2 мг/кг/сут.
В течение 1-2 месяцев может потребоваться увеличение дозы с учетом терапевтического ответа и клинической ситуации.
Побочное действие
Инфекционные и паразитарные заболевания: часто — тяжелые инфекции.
Со стороны системы кроветворения: часто — нейтропения, тромбоцитопения.
Со стороны иммунной системы: нечасто — аллергические реакции, включая анафилактическую реакцию, ангионевротический отек, крапивницу и зуд.
Со стороны нервной системы: очень часто — головная боль.
Со стороны печени и желчевыводящих путей: нечасто — повышение активности печеночных трансаминаз; частота неизвестна — неинфекционный гепатит.
Со стороны кожи и подкожных тканей: очень часто — реакции в месте инъекции; нечасто — сыпь.
Лабораторные и инструментальные данные: очень часто — повышение концентрации холестерина в крови.
Противопоказания к применению
Повышенная чувствительность к анакинре или производным белка E.coli; нейтропения (АЧН <1.5 ×109/л).
С осторожностью
Указания в анамнезе на рецидивирующие инфекции или предрасполагающие факторы, которые могут привести к развитию инфекции; повышение активности печеночных ферментов в анамнезе или печеночную недостаточность тяжелой степени тяжести; почечную недостаточность средней степени тяжести (КК 30-59 мл/мин); бронхиальную астму; злокачественные новообразования; при необходимости применения вакцинации; пациенты пожилого возраста; у пациентов, принимающих одновременно варфарин или фенитоин.
Применение при беременности и кормлении грудью
Данные о применении анакинры при беременности ограничены. Результаты исследований на животных не указывают на наличие прямой или косвенной репродуктивной токсичности. В качестве меры предосторожности рекомендуется избегать применения анакинры во время беременности, а также женщинам детородного возраста, не использующих контрацепцию.
Неизвестно, выделяется ли анакинра/метаболиты с грудным молоком у человека. Риск для новорожденного ребенка/младенца не может быть исключен. Во время лечения анакинрой грудное вскармливание следует прекратить.
Применение при нарушениях функции печени
Для пациентов с умеренной печеночной недостаточностью (класс В по классификации Чайлд-Пью) коррекция дозы не требуется. Пациентам с тяжелой печеночной недостаточностью (класс С по классификации Чайлд-Пью) следует проводить терапию с осторожностью.
Применение при нарушениях функции почек
Для пациентов с почечной недостаточностью легкой степени тяжести (КК 60-89 мл/мин) коррекция дозы не требуется. С осторожностью применять у пациентов с почечной недостаточностью средней степени тяжести (КК 30-59 мл/мин). У пациентов с почечной недостаточностью тяжелой степени тяжести (КК <30 мл/мин) и терминальной стадией почечной недостаточности, включая пациентов на диализе, следует предусмотреть введение применяемой дозы через день.
Применение у детей
Препарат разрешен для применения у детей и подростков в возрасте до 18 лет
Применение у пожилых пациентов
Данные ограничены. Предполагается, что коррекция дозы не требуется.
Особые указания
Безопасность применения анакинры у пациентов с латентным туберкулезом неизвестна. Имеются сообщения о туберкулезе у пациентов, получивших несколько курсов лечения биологическими противовоспалительными препаратами. Перед применение анакинры необходимо провести исследование на наличие латентного туберкулеза.
Другие виды противоревматической терапии ассоциировались с реактивацией гепатита В. Скрининг на вирусный гепатит должен быть выполнен в соответствии с опубликованными руководствами перед началом терапии анакинрой.
Лекарственное взаимодействие
В ходе клинических исследований у пациентов (отличных от CAPS) на фоне основной терапии метотрексатом, которые одновременно получали анакинру и этанерцепт, частота развития острых инфекций (7%) и нейтропении была выше, чем у пациентов, получавших только этанерцепт или только анакинру. Одновременный прием анакинры и этанерцепта или другого антагониста ФНОα не рекомендуется.
При хроническом воспалении образование ферментов CYP450 подавляется вследствие повышения уровня цитокинов (например, ИЛ-1). Таким образом, можно ожидать, что для антагониста рецептора ИЛ-1, такого как анакинра, образование ферментов CYP450 может быть нормализовано в ходе лечения. Это может иметь клиническое значение для субстратов CYP450 с узким терапевтическим индексом (например, варфарина и фенитоина). У пациентов, применяющих препараты данного типа, в начале или конце лечения анакинрой целесообразно проведение оценки их терапевтического эффекта, а также лабораторных показателей или концентрации этих препаратов с последующей возможной коррекцией их дозы.
Форма выпуска
Раствор для инъекций.
Упаковка
Лоток на 7 предварительно заполненных шприцов (ампул), картонная пачка.
Фармакологическое действие
Основное действующее вещество препарата Кинерет, анакинра, относится к цитокинам. Это компоненты, вырабатываемые организмом для усиления или подавления некоторых функций клеток. В случае с данным препаратом, его попадание в организм пациента позволяет снизить реакцию иммунитета на развитие ревматоидного артрита или подагры, проявляющуюся воспалением и болевыми импульсами. В результате снимаются основные симптомы заболевания.
Показания
Инъекции препарат Кинерет показаны пациентам с:
- подагрой;
- ревматоидным артритом в средней и тяжелой форме протекания.
Противопоказания
В перечень противопоказаний к назначению препарата включены такие факторы, как:
- индивидуальная непереносимость одного из компонентов препарата;
- беременность или грудное вскармливание;
- патология в почках в тяжелой форме течения;
- аллергические реакции на подобный тип препаратов в анамнезе.
Применение при беременности и кормлении грудью
Препарат противопоказан к введению в организм беременных или кормящих грудью женщин.
Особые указания
Важно убедиться в том, что препарат не подвергался замораживанию. Перед применением, раствор необходимо достать из места хранения и оставить на некоторое время для того, чтобы температура состава достигла уровня комнатной (порядка 25 градусов). После этого содержимое ампулы должно быть использовано в течение 12 часов. В противном случае, препарат необходимо утилизировать.
Препарат с большой осторожностью назначается пациентам с онкологическими заболеваниями, хронической бронхиальной астмой или инфекционными болезнями. Терапевтический процесс требует особого контроля, если у больного планируется вакцинация.
Состав
В составе раствора для инъекций значатся такие компоненты как:
- анакинра — основное активное вещество;
- цитрат, гидроксид и хлорид натрия;
- эдетат динатрия;
- полисорбат 80;
- подготовленная вода для инъекций.
Способ применения и дозы
Определением дозировки и частоты введения препарат занимается исключительно лечащий врач. Опираясь при этом на индивидуальные показатели здоровья пациента, его диагноз и степень течения болезни.
Как правило, пациентам предписывают введение содержимого одной ампулы один раз в сутки. Инъекция осуществляется строго подкожно и в одно и то же время суток.
В начале лечения введение препарата осуществляется при помощи медперсонала. Однако, пройдя соответствующий инструктаж, можно проводить процедуру самостоятельно на дому.
Побочные действия
В качестве побочных эффектов от препарата Кинерет могут выступать такие симптомы:
- отечность тканей;
- образование кровоподтеков;
- зуд и покраснение;
- головные боли;
- изменение количественных показателей лейкоцитов определенной группы в составе крови;
- повышение риска развития инфекций.
Лекарственное взаимодействие
Препарат достаточно хорошо сочетается с лекарственными средствами схожего назначения. Потому может использоваться, как часть комплексной терапии.
При этом, безопасность сочетания инъекций Кинерет с прочими медикаментами должна быть подтверждена лечащим врачом.
Действующее вещество
Анакинра.
Лекарственная форма
Раствор для инъекций.
Назначение
Назначается взрослым пациентам. Отпускается по рецепту.
Показания
Иммунодепрессивные лекарственные средства.
Условия хранения
Ампулы не замораживать. Хранить в сухом, защищенном от света месте с температурой воздуха в диапазоне 2-8 градусов по шкале Цельсия. Беречь от детей.
Применение с алкоголем
Употребление алкогольной продукции на период курса Кинерет противопоказано.
Кинерет — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер
ЛП-006746
Торговое наименование:
Кинерет®
Международное непатентованное или группировочное наименование (МНН):
Анакинра
Лекарственная форма:
раствор для подкожного введения
Состав:
В 0,67 мл раствора в одном предварительно заполненном шприце содержится:
Действующее вещество: Анакинра – 100 мг
Вспомогательные вещества: лимонная кислота, натрия хлорид, динатрия эдетата дигидрат, полисорбат-80, вода для инъекций.
Описание
Прозрачный или опалесцирующий от бесцветного до беловатого цвета раствор, который может содержать аморфные полупрозрачные или белые частицы, относящиеся к препарату
Характеристики
Анакинра представляет собой рекомбинантную версию антагониста рецептора интерлейкина-1 (IL-1ra) человека, продуцированного штаммом Е. coli с использованием технологии рекомбинантной ДНК.
Фармакотерапевтическая группа
Иммунодепрессанты: ингибиторы интерлейкина
Код АТХ
L04AC03
Фармакологические свойства
Фармакодинамика
Анакинра блокирует биологическую активность интерлейкина-1α (ИЛ-1α) и интерлейкина-1β (ИЛ-1β) путем конкурентного ингибирования их связей с рецептором интерлейкина-1 типа I. Интерлейкин-1 (ИЛ-1) является ключевым провоспалительным цитокином, медиатором многих клеточных ответов, включая те, которые имеют важное значение при синовиальном воспалении.
Анакинра ингибирует реакции, индуцированные ИЛ-1 in vitro, в том числе, индукцию оксида азота и простагландина Е2 и/или синтез коллагеназы синовиальными клетками, фибробластами и хондроцитами.
Спонтанные мутации в гене CIAS1/NLRP3 были обнаружены у большинства пациентов с криопирин-ассоциированными периодическими синдромами (CAPS). CIAS1/NLRP3 кодирует криопирин, компонент инфламмасомы. Активация инфламмасомы приводит к протеолитической матурации и секреции ИЛ-1β, который вызывает широкий спектр реакций, включая системное воспаление. У пациентов с CAPS, не получавших лечение, отмечают повышенный уровень С-реактивного белка (СРБ), амилоида А (САА) и ИЛ-6 в сыворотке крови по сравнению с нормальными уровнями. Было отмечено, что назначение препарата Кинерет® приводит к снижению уровней острофазных белков и уменьшению экспрессии ИЛ-6. Снижение уровней острофазных белков было зарегистрировано в течение первых недель лечения.
Фармакокинетика
Абсолютная биодоступность анакинры после подкожной болюсной инъекции в дозе 70 мг у здоровых субъектов (n = II) составляет 95%. Процесс абсорбции является фактором, ограничивающим скорость выведения анакинры из плазмы после подкожной инъекции.
У пациентов на фоне лечения препаратом Кинерет® максимальные концентрации анакинры в плазме обнаруживали спустя 3-7 час после подкожного введения анакинры в клинически значимых дозах (от 1 до 2 мг/кг; n = 18). Концентрация в плазме уменьшалась без очевидной фазы распределения, а период полувыведения составлял от 4 до 6 час. У пациентов, получавших препарат Кинерет®, не наблюдали непредвиденного накопления анакинры после ежедневного подкожного введения в течение до 24 недель. Средние (SD) показатели клиренса (CL/F) и объема распределения (Vd/F) по результатам популяционного фармакокинетического анализа данных 35 пациентов составили 105 (27) мл/мин и 18,5 (11) л, соответственно. Данные, полученные в исследованиях на людях и животных, показали, что почки являются основным органом, ответственным за выведение анакинры. Клиренс анакинры у пациентов, получавших препарат Кинерет®, возрастал с увеличением клиренса креатинина (КК).
Влияние демографических параметров на фармакокинетику анакинры оценивали с помощью популяционного фармакокинетического анализа данных 341 пациента, получавшего ежедневно подкожно анакинру в дозах 30, 75 и 150 мг в течение до 24 недель. Оцениваемый клиренс анакинры увеличивался с увеличением КК и массы тела. Результаты популяционного фармакокинетического анализа показали, что среднее значение плазменного клиренса после подкожной болюсной инъекции приблизительно на 14% выше у мужчин, чем у женщин, и приблизительно на 10% выше у лиц в возрасте <65 лет, чем у испытуемых лиц в возрасте 65 лет. Тем не менее, после коррекции по КК, массе тела, полу и возрасту существенных факторов, влияющих на показатель среднего плазменного клиренса, выявлено не было. Коррекция дозы препарата не требуется с учетом возраста или пола.
У пациентов с CAPS была отмечена приблизительная линейность дозы с незначительной тенденцией к более высокому, чем пропорциональному ее увеличению. Фармакокинетические данные у детей <4 лет недостаточны, но имеется клинический опыт применения у детей в возрасте от 8 месяцев. При инициации лечения в рекомендуемой суточной дозе 1-2 мг/кг проблем с безопасностью применения препарата выявлено не было. Фармакокинетические данные отсутствуют в пожилой возрастной группе пациентов с CAPS. Было продемонстрировано распределение препарата в спинномозговой жидкости.
Печеночная недостаточность
Исследование было проведено у 12 пациентов с дисфункцией печени (класс В по классификации Чайлд-Пью), получивших однократную дозу 1 мг/кг внутривенно. Фармакокинетические параметры пациентов и здоровых добровольцев значительно не различались, за исключением показателя клиренса, который у пациентов был ниже примерно на 30%. Соответствующее снижение КК было отмечено у пациентов с печеночной недостаточностью. Таким образом, снижение клиренса, наиболее вероятно, объясняется снижением функции почек у данной популяции пациентов. Эти данные подтверждают, что для пациентов с печеночной недостаточностью класса В по классификации Чайлд-Пью коррекция дозы не требуется. Для пациентов с умеренной печеночной недостаточностью коррекция дозы не требуется (класс В по классификации Чайлд-Пью). Пациентам с тяжелой печеночной недостаточностью следует проводить терапию препаратом Кинерет® с осторожностью.
Почечная недостаточность
Среднее значение плазменного клиренса на фоне лечения препаратом Кинерет® пациентов с почечной недостаточностью легкой (КК 50-80 мл/мин) и средней (КК 30-49 мл/мин) степени тяжести было снижено на 16% и 50%, соответственно. У пациентов с почечной недостаточностью тяжелой степени тяжести и терминальной стадией (КК <30 мл/мин) среднее значение клиренса плазмы было снижено на 70% и 75%, соответственно. Менее 2,5% от введенной дозы препарата Кинерет® было выведено с использованием гемодиализа или постоянного амбулаторного перитонеального диализа. Эти данные подтверждают, что для пациентов с почечной недостаточностью легкой степени тяжести (КК 50-80 мл/мин) коррекция дозы не требуется.
Показания к применению
Препарат Кинерет® показан взрослым, подросткам и детям в возрасте от 8 месяцев и старше с массой тела от 10 кг и выше для лечения криопирин-ассоциированных периодических синдромов (CAPS), включая:
- мультисистемное воспалительное заболевание неонатального возраста (NOMID) / хронический младенческий неврологический кожно-артикулярный синдром (CINCA)
- Синдром Макла-Уэллса (MWS)
- Семейный холодовой аутовоспалительный синдром (FCAS)
Противопоказания
Повышенная чувствительность к действующему веществу или любым вспомогательным веществам или производным белка E.coli.
Препарат Кинерет® не следует назначать пациентам с нейтропенией (абсолютное число нейтрофилов, ANC <1,5×109/л).
С осторожностью
- У пациентов, имеющих в анамнезе рецидивирующие инфекции или предрасполагающие факторы, которые могут привести к развитию инфекции; повышение активности печеночных ферментов в анамнезе или печеночную недостаточность тяжелой степени тяжести; почечную недостаточность средней степени тяжести (КК 30-59 мл/мин); бронхиальную астму; злокачественные новообразования, в том числе, в анамнезе (см. раздел «Особые указания»);
- У пациентов, которым требуется вакцинация (см. раздел «Особые указания»);
- У пожилых пациентов (см. раздел «Особые указания»);
- У пациентов, принимающих одновременно варфарин или фенитоин (см. раздел «Взаимодействие с другими лекарственными средствами»).
Применение при беременности и в период грудного вскармливания
Данные о применении анакинры у беременных женщин ограничены. Результаты исследований на животных не указывают на наличие прямой или косвенной репродуктивной токсичности.
В качестве меры предосторожности рекомендуется избегать применения анакинры во время беременности, а также женщинам детородного возраста, не использующих контрацепцию.
Неизвестно, проникает ли анакинра/метаболиты в грудное молоко. Риск для новорожденного ребенка/младенца не может быть исключен. Во время лечения препаратом Кинерет® грудное вскармливание следует прекратить.
Способ применения и дозы
Раствор для подкожного введения.
Лечение препаратом Кинерет® должно быть назначено врачами-специалистами, имеющими опыт в диагностике и лечении CAPS, и проводиться под их контролем.
Препарат Кинерет® вводится путем подкожной инъекции. Препарат Кинерет® поставляется готовым к применению в градуированном предварительно заполненном шприце. Градуированный предварительно заполненный шприц предназначен для дозирования от 20 до 100 мг анакинры. Поскольку минимальная доза в шприце составляет 20 мг, он не подходит для лечения детей с массой тела менее 10 кг.
Во избежание чувства дискомфорта в месте инъекции рекомендуется чередовать места инъекции. Для облегчения симптомов реакций в месте инъекции рекомендуется охлаждение места инъекции, нагревание раствора препарата до комнатной температуры, наложение на место инъекции холодных пакетов (до ее проведения и после), применение кортикостероидов и антигистаминных препаратов для местного применения.
Взрослые, подростки и дети в возрасте от 8 месяцев и старше и массой тела от 10 кг и выше
Начальная доза:
Рекомендуемая начальная доза при всех подтипах CAPS составляет 1-2 мг/кг/день в виде подкожной инъекции. Терапевтический ответ проявляется, главным образом, в виде уменьшения клинических симптомов, таких как лихорадка, сыпь, боли в суставах и головная боль, а также снижением уровней острофазных белков (СРБ/САА) или частоты обострений.
Поддерживающая доза при CAPS легкой степени тяжести (FCAS, MWS легкой степени тяжести):
Как правило, клинические и лабораторные симптомы успешно контролируются путем строгого соблюдения рекомендуемой начальной дозы (1-2 мг/кг/день).
Поддерживающая доза при CAPS тяжелой степени тяжести (MWS и NOMID/CINCA):
В течение 1-2 месяцев может потребоваться увеличение дозы с учетом терапевтического ответа. Обычная поддерживающая доза при CAPS тяжелого течения составляет 3-4 мг/кг/день, и она может быть увеличена до максимальной дозы 8 мг/кг/день.
В дополнение к оценке клинических симптомов и воспалительных маркеров при CAPS тяжелого течения, рекомендуется проводить оценку воспаления центральной нервной системы (ЦНС), в том числе внутреннего уха (с помощью магниторезонансной томографии (MPT) или компьютерной томографии (КТ), люмбальной пункции и аудиологического исследования) и органов зрения (офтальмологическая оценка) после первоначального 3-месячного курса лечения, а затем каждые 6 месяцев, до тех пор, пока не будет определен эффективный режим терапии. Когда контроль состояния пациента успешно достигнут, мониторинг клинических, офтальмологических показателей и параметров ЦНС можно проводить ежегодно.
Применение у пожилых пациентов (≥65 лет)
Данные о применении препарата у пожилых пациентов с CAPS ограничены. Предположительно, коррекция дозы препарата не требуется.
Применение у детей (в возрасте <18 лет)
Данные о применении препарата у детей в возрасте до восьми месяцев отсутствуют.
Режим дозирования и способ применения у детей в возрасте 8 месяцев и старше с массой тела 10 кг и выше соответствует режиму применения у взрослых пациентов с CAPS и зависит от массы тела.
Применение у пациентов с нарушением функции печени
Для пациентов с умеренной печеночной недостаточностью (класс В по классификации Чайлд-Пью) коррекция дозы не требуется. Пациентам с тяжелой печеночной недостаточностью (класс С по классификации Чайлд-Пью) следует проводить терапию препаратом Кинерет® с осторожностью.
Применение у пациентов с нарушением функции почек
Для пациентов с почечной недостаточностью легкой степени тяжести (КК 60-89 мл/мин) коррекция дозы не требуется. Препарат Кинерет® следует применять с осторожностью у пациентов с почечной недостаточностью средней степени тяжести (КК 30-59 мл/мин). У пациентов с почечной недостаточностью тяжелой степени тяжести (КК <30 мл/мин) и терминальной стадией почечной недостаточности, включая пациентов на диализе, следует предусмотреть введение назначенной дозы препарата Кинерет® через день.
Если препарат Кинерет® введен в большей дозе, чем это было необходимо
При случайном введении препарата Кинерет® в дозе, превышающей необходимую, не должно возникнуть серьезных проблем. Тем не менее, в подобном случае пациенту следует обратиться к врачу. В любом случае, если пациент почувствовал себя нехорошо, он/она должен/должна незамедлительно обратиться к врачу.
Если пропущено введение дозы препарата Кинерет®
Если пропущено введение дозы препарата Кинерет®, пациенту следует обратиться к врачу, чтобы обсудить, когда следует вводить следующую дозу.
Инструкция по подготовке и выполнению инъекции препарата Кинерет®
Данный раздел содержит информацию о том, как сделать себе или своему ребенку инъекцию препарата Кинерет®. Не следует вводить препарат Кинерет® себе или своему ребенку, если Вы не прошли инструктаж у врача или медицинской сестры. Если у вас есть вопросы о том, как вводить препарат, пожалуйста, обратитесь за помощью к врачу или медицинской сестре.
Препарат Кинерет® представляет собой стерильный раствор, не содержащий консервантов. Только для однократного применения.
Шприц не следует встряхивать. Перед инъекцией необходимо дать нагреться шприцу до комнатной температуры.
Перед введением нужно проверить внешний вид раствор на наличие механических включений и изменения цвета. Вводить можно только прозрачный или слабо опалесцирующий от бесцветного до белесого цвета раствор, который может содержать аморфные полупрозрачные или белые частицы, относящиеся к препарату.
Присутствие таких частиц не влияет на качество препарата.
Шприц предназначен только для одноразового применения. Любые неиспользованные остатки препарата следует утилизировать.
Как сделать инъекцию самостоятельно или с помощью другого лица с использованием препарата Кинерет® в градуированном предварительно заполненном шприце
Вводить препарат себе или своему ребенку необходимо в одно и то же время каждый день. Поскольку препарат Кинерет® предназначен для подкожных инъекций, его вводят только под кожу.
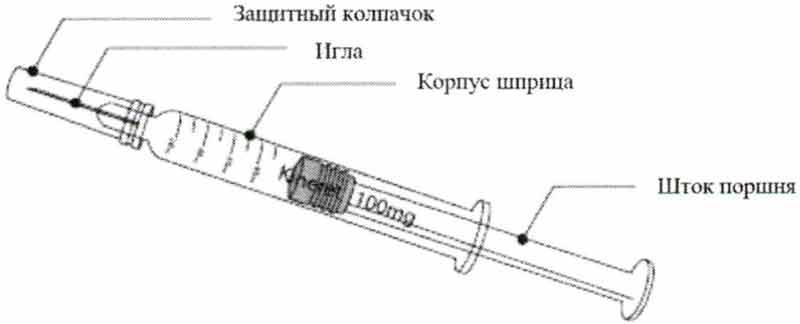
Для выполнения подкожной инъекции препарата себе или своему ребенку вам понадобятся:
- Препарат Кинерет® в градуированном предварительно заполненном шприце;
- Спиртовые салфетки или аналоги;
- Стерильная марля или ткань.
Что вам следует сделать перед подкожным введением препарата Кинерет® себе или своему ребенку?
- Достаньте препарат Кинерет® в градуированном предварительно заполненном шприце из холодильника.
- Не встряхивайте шприц.
- Проверьте дату истечения срока годности на этикетке предварительно заполненного шприца (в редакции «До:»). Не используйте препарат Кинерет® после истечения срока годности, указанного на упаковке.
- Проверьте внешний вид раствора препарата Кинерет® в шприце. Раствор должен быть прозрачным или слабо опалесцирующим, от бесцветного до белого цвета. Раствор может содержать аморфные полупрозрачные или белые частицы, относящиеся к препарату. Присутствие этих частиц не влияет на качество препарата. Раствор не следует вводить, если он изменил цвет или стал непрозрачным, или если имеются какие-либо другие частицы, отличные от прозрачно-белых частиц.
- Для облегчения симптомов реакций в месте инъекции препарата оставьте шприц при комнатной температуре в течение приблизительно 30 минут или осторожно подержите его в руке в течение нескольких минут. Не нагревайте Кинерет® каким-либо иным способом (например,не нагревайте его в микроволновой печи или при помощи горячей воды).
- Не снимайте защитный колпачок со шприца, если Вы не готовы вводить препарат.
- Тщательно вымойте руки.
- Найдите удобную, хорошо освещенную и чистую поверхность и поместите все необходимое оборудование перед собой.
- Убедитесь, что Вы знаете, какую дозу препарата Кинерет® назначил ваш врач; от 20 до 90 мг, 100 мг или выше.
• Если врач назначил дозу 100 мг, смотрите раздел «Как приготовить дозу 100 мг».
• Если врач назначил более низкую дозу, смотрите раздел «Как приготовить дозу от 20 до 90 мг».
Как приготовить дозу 100 мг
Перед введением препарата Кинерет® вам следует выполнить следующее:
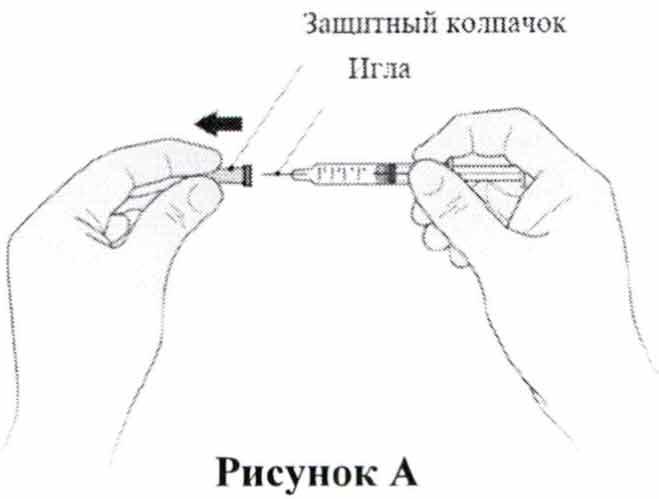
- Возьмите шприц и осторожно снимите с иглы защитный колпачок, не вращая его.
Потяните колпачок прямо, как показано на Рис. А. Не прикасайтесь к игле и не нажимайте на поршень. Сразу выбросьте колпачок от иглы.
- Вы можете заметить наличие небольшого пузырька воздуха в шприце. Вам не следует удалять пузырьки воздуха перед инъекцией. Введение раствора с пузырьком воздуха безопасно.
- Теперь Вы можете использовать предварительно заполненный шприц согласно описания в следующих разделах «Куда следует делать себе инъекцию?» и «Как сделать себе инъекцию?».
Как приготовить дозу от 20 до 90 мг
Перед введением препарата Кинерет® вам следует выполнить следующее:
- Возьмите шприц и осторожно снимите с иглы колпачок, не вращая его. Потяните колпачок прямо, как показано на Рис. А. Не прикасайтесь к игле и не нажимайте на поршень. Сразу выбросьте колпачок от иглы.
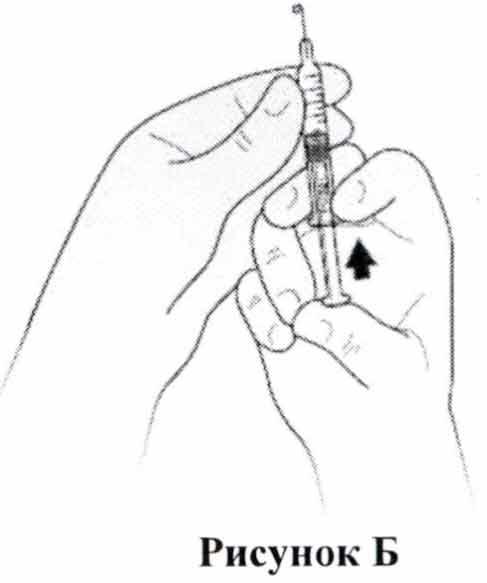
- Вам следует держать шприц в одной руке, направляя иглу прямо вверх, как показано на Рис. Б. Поместите и держите большой палец на штоке поршня до тех пор, пока на кончике иглы не появится крошечная капля жидкости.
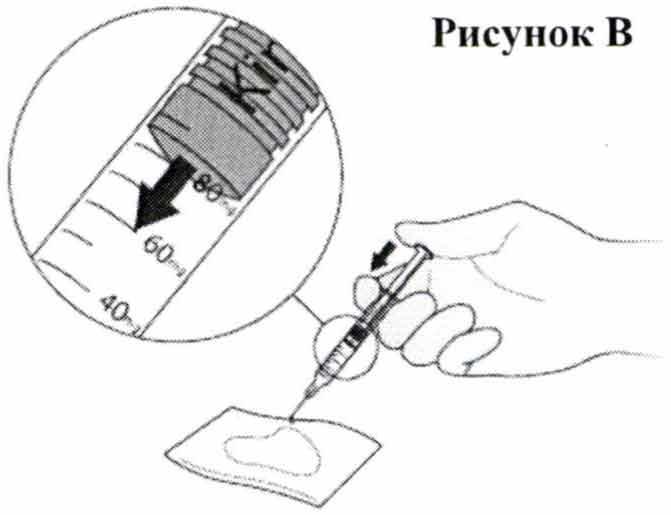
- Переверните шприц таким образом, чтобы игла теперь была направлена вниз. Поместите стерильную марлю или ткань на плоскую поверхность и держите над ней шприц с иглой, направленной в сторону марли или ткани, как показано на Рис. В. Убедитесь в том, что игла не касается марли или ткани.
- Поместите большой палец на шток поршня и нажимайте на него до тех пор, пока передняя часть поршня не дойдет до метки на шкале, указывающей на необходимую вам дозу препарата Кинерет® (ваш врач проинформирует вас о назначенной дозе препарата). Выпущенная струя жидкости впитается марлей или тканью, как показано на Рис. В.
- Если вы не смогли установить правильную дозу, утилизируйте шприц и используйте новый.
- Теперь, вы можете использовать предварительно заполненный шприц согласно описанию в следующих разделах «Куда следует делать себе инъекцию?» и Как сделать себе инъекцию?».
Куда следует делать себе инъекцию?
Наиболее подходящие участки для выполнения инъекции себе или своему ребенку (смотрите Рис. Г):
- живот (за исключением области вокруг пупка)
- верхняя часть бедра
- верхние наружные области ягодиц;
- наружная область плеча
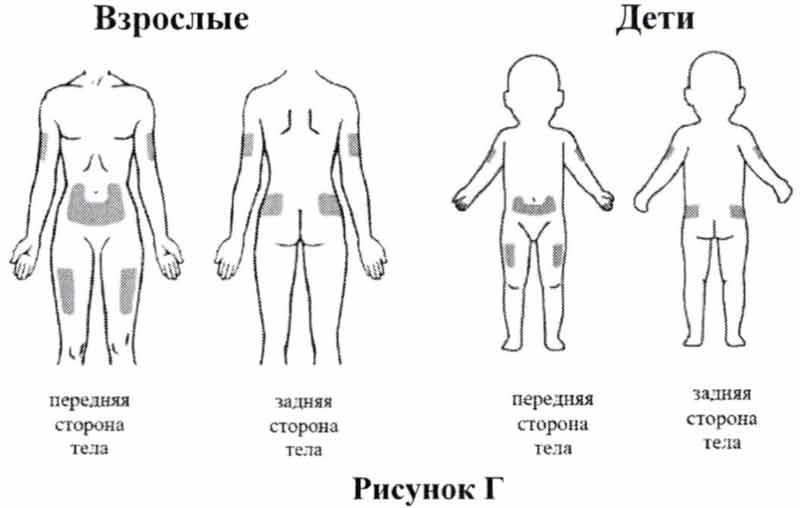
Каждый раз меняйте место инъекции, чтобы избежать развития болезненности в одной области. При введении препарата с посторонней помощью может быть задействована задняя поверхность вашего плеча.
Как сделать себе инъекцию?
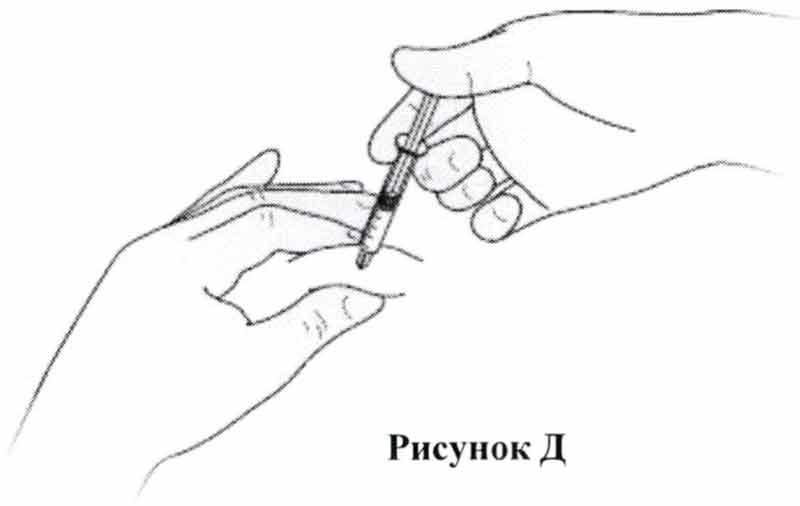
- Продезинфицируйте кожу с помощью спиртовых салфеток и зажмите кожу между большим и указательным пальцами, не сдавливая ее.
- Введите иглу в кожу полностью, как было вам показано медицинской сестрой или врачом.
- Вводите раствор препарата медленно и равномерно в образовавшуюся кожную складку, как показано на Рис. Д.
- После введения препарата, извлеките иглу и отпустите кожу.
- Любые неиспользованные остатки препарата должны быть утилизированы. Используйте один шприц только для одной инъекции. Не используйте шприц повторно, поскольку это может стать причиной развития инфекции.
Запомните
Если у вас есть какие-либо проблемы, пожалуйста, незамедлительно обращайтесь к своему врачу или медицинской сестре за помощью и советом.
Утилизация использованных шприцев и материалов
- Не надевайте колпачок на использованные иглы
- Держите использованные шприцы в недоступном для детей месте
- Никогда не кладите использованный шприцы в обычное мусорное ведро с бытовыми отходами
- Если вам нужна доза препарата менее 100 мг, как было сказано выше, следует удалить избыток препарата из шприца на марлю или ткань. После инъекции утилизируйте мокрую марлю или ткань с вашим шприцем и очистите поверхность чистой тканью
- Использованные шприцы и марлю или ткань следует утилизировать согласно местным законодательным актам. Данные меры помогут защитить окружающую среду.
Побочное действие
Краткая характеристика профиля безопасности
Данные о нежелательных лекарственных реакциях (НЛР) у пациентов с CAPS получены в открытом КИ у 43 пациентов с NOMID/CINCA на фоне терапии препаратом Кинерет®, длительностью до 5 лет и показателем общей экспозиции 159,8 пациенто-лет. В ходе 5-летнего КИ у 14 пациентов (32,6%) было зарегистрировано 24 серьезных нежелательных явления (СНЯ). Одиннадцать СНЯ у 4 пациентов (9,3%) были расценены как связанные с применением препарата Кинерет®. Ни у одного пациента лечение препаратом Кинерет® не было прекращено из-за НЛР.
Ниже представлены НЛР, которые классифицированы в соответствии с поражением органов и систем органов (медицинский словарь для нормативно-правовой деятельности MedDRA) и частотой возникновения.
| Класс системы органов | Частота | Нежелательная лекарственная реакция |
| Инфекционные и паразитарные заболевания | Часто (от ≥1/100 до <1/10) | Тяжелые инфекции |
| Нарушения со стороны кровеносной и лимфатической систем | Часто (от ≥1/100 до <1/10) | Нейтропения Тромбоцитопения |
| Нарушения со стороны иммунной системы | Нечасто (от ≥1/1000 до <1/100) | Аллергические реакции, включая анафилактическую реакцию, ангионевротический отек, крапивницу и зуд |
| Нарушения со стороны нервной системы | Очень часто (≥1/10) | Головная боль |
| Нарушения со стороны печени и желчевыводящих путей | Нечасто (от ≥1/1000 до <1/100) | Повышение печеночных трансаминаз |
| Частота неизвестна (не может быть идентифицирована по имеющимся данным) | Неинфекционный гепатит | |
| Нарушения со стороны кожи и подкожных тканей | Очень часто (≥1/10) | Реакции в месте инъекции |
| Нечасто (от ≥1/1000 до <1/100) | Сыпь | |
| Лабораторные и инструментальные данные | Очень часто (≥1/10) | Повышение холестерина в крови |
Если любые из указанных в инструкции побочных эффектов усугубляются, или пациент заметил любые другие побочные эффекты, не указанные в инструкции, следует сообщить об этом врачу.
Тяжелые инфекции
У 43 пациентов с CAPS за 5-летний период наблюдения частота тяжелых инфекций составляла 0,1/год, из которых чаще всего встречалась пневмония и гастроэнтерит. Применение препарата Кинерет® было временно прекращено у одного пациента; остальные пациенты продолжили лечение препаратом Кинерет® во время течения инфекций.
В ходе КИ и пострегистрационного применения препарата были отмечены редкие случаи инфекций условно-патогенными организмами, которые включали грибковые, микобактериальные, бактериальные и вирусные патогены. Инфекции были отмечены во всех системах органов и были зарегистрированы у пациентов, получавших препарат Кинерет® отдельно или в сочетании с иммунодепрессантами.
Нейтропения
В плацебо-контролируемых КИ препарата Кинерет® для отличного от CAPS состояния в ходе лечения было отмечено незначительное уменьшение средних значений общего числа лейкоцитов и абсолютного числа нейтрофилов. Нейтропения (ANC <1,5×109/л) была зарегистрирована у 2,4% пациентов, получавших препарат Кинерет® по сравнению с 0,4% пациентов, получавших плацебо. Ни у одного из этих пациентов не было диагностировано острых инфекций, связанных с нейтропенией. Из 43 пациентов с CAPS, наблюдавшихся в течение пяти лет, нейтропения была отмечена у 2 пациентов. В обоих случаях нейтропения разрешилась в ходе продолжения лечения препаратом Кинерет®.
Тромбоцитопения
В ходе КИ препарата Кинерет®, назначаемого по показаниям, не включающим CAPS, тромбоцитопения была зарегистрирована у 1,9% пациентов в группе препарата Кинерет® по сравнению с 0,3% пациентов в группе плацебо. Тромбоцитопения была легкой (количество тромбоцитов >75×109/л). У пациентов с CAPS также наблюдали легкую тромбоцитопению. В ходе пострегистрационного применения препарата Кинерет® была зарегистрирована тромбоцитопения, в том числе, эпизодически отмечали случаи тяжелой тромбоцитопении (количество тромбоцитов <10×109/л).
Злокачественные опухоли
В рамках КИ препарата Кинерет®, назначаемого по показаниям, не включающим CAPS, общая частота случаев злокачественных опухолей была одинаковой в группе пациентов, принимавших препарат Кинерет®, и в группе, принимавшей плацебо, и не отличалась от частоты случаев, зарегистрированных в целом в исследуемых популяциях. Кроме того, общая частота случаев злокачественных опухолей не увеличивалась на протяжении трех лет применения препарата Кинерет®.
Аллергические реакции
Аллергические реакции на препарат Кинерет®, включая анафилактические реакции, ангионевротический отек, крапивницу, сыпь и зуд, встречались нечасто. Большинство из этих реакций были представлены макулопапулезной или уртикарной сыпью.
У 43 пациентов с CAPS, наблюдавшихся в течение пяти лет, не было серьезных аллергических реакций и случаев, требующих досрочного прекращения лечения препаратом Кинерет®.
Иммуногенность
В ходе КИ у большинства пациентов с CAPS были обнаружены антитела к анакинре. Не было отмечено клинически значимого влияния наличия антител к анакинре на фармакокинетику, эффективность и безопасность препарата Кинерет®.
Изменения функции печени
В ходе КИ наблюдали случаи транзиторного повышения активности печеночных ферментов. Это повышение не было связано с признаками или симптомами повреждения клеток печени. В ходе пострегистрационного применения препарата Кинерет® были получены сообщения о единичных случаях неинфекционного гепатита.
В пострегистрационном периоде изменения функции печени были отмечены преимущественно у пациентов с предрасполагающими факторами в анамнезе, например, имевших повышенную активность трансаминаз до назначения препарата Кинерет®.
Реакции в месте инъекции
В ходе КИ препарата Кинерет®, назначаемого по показаниям, отличным от CAPS, наиболее частыми НЛР являлись реакции в месте инъекции. Большинство (95%) реакций в месте инъекций были от легкой до умеренной степени тяжести. Реакции в месте инъекции обычно характеризовались одним или несколькими признаками, такими как: эритема, экхимоз, воспаление и боль. Из 43 пациентов с CAPS, наблюдавшихся в течение пяти лет, ни один пациент не прекратил лечение препаратом Кинерет® и не приостановил его временно в связи с реакциями в месте инъекции. Реакции в месте инъекции обычно возникают в течение двух недель с момента введения препарата, и исчезают в течение 4-6 недель. У пациентов, у которых ранее не регистрировали реакций в месте инъекции, эти реакции редко развивались после первого месяца терапии.
Повышение холестерина в крови
У 775 пациентов, которые в рамках КИ по отличным от CAPS показаниям получали препарат Кинерет® в суточных дозах 30 мг, 75 мг, 150 мг, I мг/кг или 2 мг/кг, наблюдали увеличение общего содержания холестерина от 2,4% до 5,3% через 2 недели после начала лечения препаратом Кинерет® вне зависимости от дозы препарата. Подобные изменения показателей общего холестерина отмечали и через 24 недели лечения препаратом Кинерет®. Лечение плацебо (n = 213) сопровождалось уменьшением общего содержания холестерина приблизительно на 2,2% на неделе 2 и на 2,3% на неделе 24. Данных о липопротеинах низкой плотности и липопротеинах высокой плотности не имеется.
Применение у детей
Препарат Кинерет® изучали у пациентов в возрасте от 8 месяцев до <18 лет (36 пациентов с CAPS и 86 пациентов с другими состояниями) в течение пяти лет. За исключением инфекций и связанных с ними симптомов, которые чаще отмечались у пациентов в возрасте <2 лет, профиль безопасности был сходным у детей всех возрастных групп. Педиатрическая и взрослая популяции пациентов имели сходный профиль безопасности; новых клинически значимых НЛР отмечено не было.
Передозировка
В ходе КИ не было отмечено ограничивающей дозу токсичности. В рамках КИ по отличным от CAPS показаниям 1015 пациентов получали препарат Кинерет® внутривенно в дозах до 2 мг/кг/час на протяжении 72 часов. Профиль нежелательных явлений в этих КИ не отличался от профиля безопасности в КИ, где применяли более низкие дозы препарата Кинерет®.
Взаимодействие с другими лекарственными средствами
Взаимодействие между препаратом Кинерет® и другими лекарственными средствами направленно не изучали. В рамках КИ не было отмечено взаимодействия между препаратом Кинерет® и другими лекарственными средствами (в том числе нестероидными противовоспалительными препаратами, глюкокортикостероидами и базисными противоревматическими препаратами (БПРП)).
В ходе КИ у пациентов (отличных от CAPS) на фоне основной терапии метотрексатом, которые одновременно получали препарат Кинерет® и этанерцепт, частота развития острых инфекций (7%) и нейтропении была выше, чем у пациентов, получавших только этанерцепт или только препарат Кинерет®. Одновременный прием препарата Кинерет® и этанерцепта или другого антагониста фактора некроза опухоли-альфа (ФНО-α) не рекомендуется.
При хроническом воспалении образование ферментов CYP450 подавляется вследствие повышения уровня цитокинов (например, ИЛ-1). Таким образом, можно ожидать, что для антагониста рецептора ИЛ-1, такого как анакинра, образование ферментов CYP450 может быть нормализовано в ходе лечения. Это может иметь клиническое значение для субстратов CYP450 с узким терапевтическим индексом (например, варфарина и фенитоина). У пациентов, применяющих препараты данного типа, в начале или конце лечения препаратом Кинерет® целесообразно проведение оценки их терапевтического эффекта, а также лабораторных показателей или концентрации этих препаратов с последующей возможной коррекцией их дозы.
Особые указания
Аллергические реакции
Аллергические реакции, включая анафилактические реакции и ангионевротический отек, встречались нечасто. Чаще всего отмечали макулопапулезную или уртикарную сыпь. При появлении признаков тяжелой аллергической реакции введение препарата Кинерет® следует прекратить и назначить надлежащее лечение.
Нежелательные явления со стороны печени
В ходе КИ были отмечены случаи временного повышения активности печеночных ферментов. Эти повышения не были связаны с признаками или симптомами повреждения клеток печени. В ходе пострегистрационного применения препарата сообщалось о нежелательных явлениях со стороны печени, не оказывавших влияния на ее функцию. У большинства пациентов были выявлены предрасполагающие факторы, например, случаи повышения активности печеночных трансаминаз в анамнезе. Оценку эффективности и безопасности применения препарата Кинерет® у пациентов с активностью аспартатаминотрансферазы/аланинаминотрансферазы ≥1,5 верхнего уровня нормы не проводили.
Тяжелые инфекции
В ходе КИ препарата Кинерет® при показаниях, отличных от CAPS, была отмечена взаимосвязь применения препарата Кинерет® с увеличением заболеваемости тяжелыми инфекциями (1,8%) по сравнению с плацебо (0,7%). Для небольшого числа пациентов с бронхиальной астмой, заболеваемость острыми инфекциями была выше у пациентов, получавших препарат Кинерет® (4,5%), по сравнению с пациентами, получавшими плацебо (0%); в основном были диагностированы инфекции респираторного тракта. Оценку безопасности и эффективности применения препарата Кинерет® у пациентов с хроническими и острыми инфекциями не проводили.
Препарат Кинерет® не следует назначать пациентам с активными инфекциями. Для пациентов с CAPS существует риск обострения заболевания после прекращения терапии препаратом Кинерет®. При условии тщательного контроля лечение препаратом Кинерет® может быть продолжено и во время тяжелой инфекции.
Врачи должны соблюдать осторожность при введении препарата Кинерет® пациентам, имеющим в анамнезе рецидивирующие инфекции или предрасполагающие факторы, которые могут привести к развитию инфекции.
Безопасность применения препарата Кинерет® у пациентов с латентным туберкулезом неизвестна. Имеются сообщения о туберкулезе у пациентов, получивших несколько курсов лечения биологическими противовоспалительными препаратами. Перед назначением препарата Кинерет® пациентов необходимо проверять на наличие латентного туберкулеза. Имеющиеся медицинские рекомендации также должны быть приняты во внимание.
Другие виды противоревматической терапии ассоциировались с реактивацией гепатита В. Скрининг на вирусный гепатит должен быть выполнен в соответствии с опубликованными руководствами перед началом терапии препаратом Кинерет®.
Почечная недостаточность
Препарат Кинерет® выводится путем клубочковой фильтрации и последующего метаболизма в почечных канальцах. Соответственно, плазменный клиренс препарата Кинерет® снижается при ухудшении функции почек.
Для пациентов с почечной недостаточностью легкой степени тяжести (КК 60-89 мл/мин) коррекция дозы не требуется. У пациентов с почечной недостаточностью средней степени тяжести (КК 30 – 59 мл/мин) препарат Кинерет® следует применять с осторожностью. У пациентов с тяжелой почечной недостаточностью (КК <30 мл/мин) или терминальной почечной недостаточностью, включая пациентов на диализе, следует предусмотреть введение назначенной дозы препарата Кинерет® через день.
Нейтропения
Применение препарата Кинерет® было, как правило, связано с нейтропенией (ANC <1,5×109/л) в плацебо-контролируемых КИ при другом показании, но случаи нейтропении также были отмечены у пациентов с CAPS. Терапию препаратом Кинерет® не следует начинать у пациентов с нейтропенией (ANC <1,5×109/л). Рекомендуется проводить оценку количества нейтрофилов до начала лечения и во время применения препарата Кинерет® ежемесячно в течение первых 6 месяцев лечения и далее – каждые 3 месяца. Пациентам, у которых развивается нейтропения (ANC <1,5×109/л), следует проводить тщательный контроль количества нейтрофилов, и лечение препаратом Кинерет® у них должно быть прекращено. Оценку безопасности и эффективности применения препарата Кинерет® у пациентов с нейтропенией не проводили.
Иммуносупрессия
Влияние лечения препаратом Кинерет® на уже имеющуюся злокачественную опухоль не изучали. Поэтому, не рекомендуется назначать препарат Кинерет® пациентам с уже имеющимися злокачественными опухолями.
Вакцинация
Данные плацебо-контролируемого КИ (n = 126) не показали различий в выработке антител к столбняку в ответ на введение дифтерийно-столбнячной вакцины пациентам, получавшим препарат Кинерет® и плацебо. Данные о результатах вакцинации при введении других инактивированных антигенов пациентам на фоне лечения препаратом Кинерет® отсутствуют.
Нет данных и о реакциях на применение живых вакцин или о вторичной передаче инфекции с живыми вакцинами пациентам, получающим Кинерет®. Таким образом, живые вакцины не следует назначать пациентам на фоне лечения препаратом Кинерет®.
Применение у пожилых пациентов (≥65 лет)
В рамках КИ по показаниям, отличным от CAPS, препарат Кинерет® изучали у 752 пациентов в возрасте ≥ 65 лет, в том числе, у 163 пациентов в возрасте ≥ 75 лет. В целом, не было обнаружено различий в безопасности и эффективности препарата у пациентов пожилого и более молодого возраста. Опыт лечения пожилых пациентов с CAPS ограничен. Поскольку у пожилых заболеваемость инфекциями более высокая, во время лечения этих пациентов следует соблюдать осторожность.
Вспомогательные вещества
Данный препарат содержит менее 1 ммоль натрия (23 мг) на каждые 100 мг воды, что практически означает отсутствие натрия в препарате.
Влияние на способность управлять автотранспортными средствами, механизмами
Препарат Кинерет® не оказывает влияния на способность управлять транспортными средствами и работу с механизмами, требующими повышенной концентрации внимания.
Форма выпуска
Раствор для подкожного введения, 150 мг/мл
0,67 мл раствора (100 мг) в градуированном предварительно заполненном шприце, состоящем из стеклянного цилиндра (боросиликатное стекло I типа USP) со встроенной нержавеющей стальной иглой для подкожного введения (размер 29G), закрытой эластомерным колпачком с дополнительным внешним жестким полипропиленовым колпачком, поршня и плунжера.
7 градуированных предварительно заполненных шприцев помещают в ПВХ блистер (поддон). 1 блистер (поддон) с вместе с инструкцией по применению помещают в картонную коробку (вторичную упаковку). 4 картонные коробки, содержащие по 7 шприцев, упакованы в картонную пачку (третичную упаковку) (всего в картонной пачке 28 градуированных предварительно заполненных шприцев).
На каждую картонную пачку наклеивают прозрачную защитную этикетку для контроля первого вскрытия.
Условия хранения
Хранить при температуре от 2 до 8 °С в картонной пачке для защиты от воздействия света.
Не замораживать.
Храните в местах, недоступных для детей.
Препарат Кинерет® допустимо хранить в течение 12 часов при температуре не выше 25°С. По истечению этого периода препарат необходимо выбросить.
Срок годности
3 года.
Не использовать после истечения срока годности.
Условия отпуска
Отпускают по рецепту.
Владелец регистрационного удостоверения и выпускающий контроль качества:
Сведиш Орфан Биовитрум АБ (пабл) / Swedish Orphan Biovitrum АВ (publ)
SE 112 76 Стокгольм, Швеция / SE 11276 Stockholm, Sweden
Производитель и первичный упаковщик:
Патеон Италия С.П.А. / Patheon Italia S.P.A.
Виале Дж.Б. Стукки, 110 20900 Монца (Монца-Брианца), Италия / Viale G. В. Stukki, 110 20900 Monza (Monza-Brianza), Italy
Адрес для направления претензий потребителей:
ООО «Сведиш Орфан Биовитрум»
С/О Шведский Торговый Совет
119034, Москва, ул. Пречистенка 40/2, дом 1,4 подъезд, 2 этаж
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Ингибиторы фактора некроза опухоли альфа (ФНОα).
Код ATX: L04AB02
Инфлимаб – это лекарственное средство, которое представляет собой генно-инженерное (мышино-человеческое) моноклональное антитело к особому белку, активно участвующему в воспалительных и иммунных реакциях – фактору некроза опухоли альфа (ФНОα). Инфлимаб подавляет биологические реакции, связанные с избыточным образованием ФНОα и его повреждающим действием на организм пациента при аутоиммунных заболеваниях.
Он применяется для лечения:
Болезнь Крона (1.1): с целью уменьшение симптомов, а также перевода заболевания в стадию клинической ремиссии (значительное или полное исчезновение проявлений заболевания и, соответственно, жалоб) и поддержание такого состояния у взрослых пациентов с активным заболеванием средней и тяжелой степени тяжести, у которых проведенная стандартная терапия была неэффективной.
Уменьшение числа свищей и поддержание их в зарубцевавшемся состоянии у взрослых пациентов со свищевой формой заболевания.
Болезнь Крона у детей (1.2): уменьшение проявлений заболевания, а также перевод заболевания в стабильное состояние и его поддержание у пациентов детского возраста с активным заболеванием средней и тяжелой степени, у которых ранее проведенная стандартная терапия была неэффективной.
Язвенный колит (1.3): уменьшение симптомов, а также перевод заболевания в стабильное состояние и его поддержание, заживление слизистой оболочки кишечника, а также для отмены глюкокортикостероидов у взрослых пациентов с активным заболеванием средней и тяжелой степени тяжести, у которых ранее проведенная стандартная терапия была неэффективной.
Язвенный колит у детей (1.4): уменьшение симптомов, а также перевод заболевания в стабильное состояние и его поддержание у пациентов детского возраста с активным заболеванием средней и тяжелой степени тяжести, у которых ранее проведенная стандартная терапия была неэффективной.
Ревматоидный артрит (1.5) в комбинации с метотрексатом: уменьшение симптомов, замедление прогрессирования повреждения суставов и улучшение функционального состояния у пациентов с активным заболеванием средней и тяжелой степени тяжести.
Анкилозирующий спондилит (1.6): уменьшение симптомов у пациентов с активным заболеванием.
Псориатический артрит (1.7): уменьшение симптомов активного артрита, замедление прогрессирования повреждения суставов и улучшение функционального состояния.
Бляшковидный псориаз (1.8): лечение взрослых пациентов с тяжёлым течением хронического бляшковидного псориаза (т.е. распространенным и инвалидизирующим), которым показана системная терапия.
Активное вещество данного лекарственного средства быстро образует стабильные комплексы с человеческим ФНОα, что сопровождается снижением биологической активности ФНОα. Это приводит к уменьшению проникновения иммунных клеток в очаги воспаления, и, следовательно, к уменьшению разрушения тканей.
Лекарственное средство Инфлимаб в некоторых случаях могут назначать совместно с лекарственным средством метотрексат.
Если планируется параллельный прием каких-либо других лекарственных средств, внимательно изучите листки-вкладыши к упаковкам данных лекарственных средств. При возникновении любых вопросов относительно приема и комбинирования лекарственных средств обратитесь к Вашему лечащему врачу или фармацевту.
1. Использование инфликсимаба в дозе более 5 мг/кг при наличии хронической сердечной недостаточности средней и тяжелой степени тяжести.
2. Наличие в прошлом тяжелых аллергических реакций (реакции гиперчувствительности) на Инфлимаб или известная гиперчувствительность на вспомогательные вещества Инфлимаба либо на любые мышиные белки.
Не принимайте Инфлимаб, если:
• у Вас имеется гиперчувствительность к активным или к любому из вспомогательных веществ (перечислены ниже), входящих в состав данного лекарственного средства;
Если этот пункт относится к Вам, откажитесь от применения Инфлимаба и немедленно сообщите об этом своему лечащему врачу.
Ваш лечащий врач должен знать, имеются ли у Вас нижеперечисленные проблемы, до начала лечения Инфлимабом:
1. Тяжелые инфекции: не использовать Инфлимаб во время активного инфекционного процесса. При развитии инфекционного процесса необходимо постоянно тщательно оценивать ситуацию и прекратить использование Инфлимаба при утяжелении инфекционного процесса.
2. Грибковые инфекции: повышен риск развития не только местных, но и тяжелых распространенных грибковых инфекции у пациентов во время терапии Инфлимабом. Требуется тщательное клиническое наблюдение, и при подозрении на грибковое поражение – консультация специалиста и раннее начало противогрибковой терапии. Для пациентов, проживающих или путешествующих в регионах, где распространены особые грибковые заболевания – тщательно взвесить соотношение риск/польза до начала терапии лекарственным средством Инфлимаб.
3. Злокачественные новообразования: риск развития онкологических заболеваний, включая лимфому, повышается у пациентов, находящихся на лечении Инфлимабом по сравнению с контрольной группой. В связи с риском развития Т-клеточной лимфомы печени и селезенки должно быть внимательно оценено соотношение риск/польза, особенно у пациентов мужского пола с болезнью Крона или язвенным колитом, которые получают терапию азатиоприном или 6-меркаптопурином.
4. Реактивация вируса гепатита В: Вам необходимо сдать анализ крови на наличие вируса гепатита В перед началом лечения Инфлимабом. В случае выявления носительства вируса гепатита В рекомендовано постоянное тщательное наблюдение на протяжении всего срока терапии и в течение нескольких месяцев после завершения лечения Инфлимабом. Если возникает реактивация гепатита В, необходимо прекратить прием Инфлимаба и начать противовирусную терапию.
5. Гепатотоксичность: описаны редкие случаи тяжелых реакций со стороны печени, иногда с летальным исходом или требующих трансплантации печени. В случае развития желтухи и/или повышении уровня печеночных ферментов необходимо прекратить терапию Инфлимабом.
6. Хроническая сердечная недостаточность: впервые выявленная или ухудшение симптомов.
7. Гематологические реакции: рекомендовано немедленно обратиться за медицинской помощью в случае развития симптомов гематологических реакций (длительно существующая лихорадка, кровоподтеки, кровотечения, бледность) или выявления снижения количества клеток крови по данным лабораторных исследований для рассмотрения необходимости прекращения терапии Инфлимабом.
8. Гиперчувствительность: при введении лекарственного средства Инфлимаб может развиться тяжелая инфузионная реакция, в том числе анафилактический шок или реакция по типу сывороточной болезни.
9. Демиелинизирующие заболевания центральной или периферической нервной системы: ухудшение течения имеющегося заболевания или впервые выявленное.
10. Волчаночно-подобный синдром (лихорадка, боли в мышцах и суставах, артрит, накопление жидкости в полостях тела, реже – анемия): следует прекратить терапию Инфлимабом в случае развития данного синдрома.
11. Живые вакцины или лекарственные средства, содержащие инфекционные агенты: не должны применяться совместно с Инфлимабом. Необходимо провести вакцинацию пациентов детского возраста согласно календарю прививок (по возможности, до начала лечения Инфлимабом).
Дети и подростки
Инфлимаб не изучался среди детей с болезнью Крона или язвенным колитом в возрасте до 6 лет.
Проинформируйте своего врача или фармацевта о параллельном приеме других лекарственных средств, а также если Вы принимали лекарственные средства незадолго до приема Инфлимаба или собираетесь принимать по завершении лечения Инфлимабом.
Совместное применение лекарственного средства Инфлимаб с анакинрой и абатацептом увеличивает риск тяжелых инфекций.
Беременность
Если Вы беременны или кормите грудью, или думаете, что забеременели, или планируете беременность, то перед началом применения лекарственного средства Инфлимаб проконсультируйтесь с Вашим лечащим врачом.
Инфлимаб может оказывать незначительное влияние на способность управлять автомобилем и работать с механизмами. Следует соблюдать осторожность при управлении транспортными средствами и занятии другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций, так как лекарственное средство может вызывать головокружение и другие нежелательные реакции, которые могут влиять на эти способности.
Всегда принимайте лекарственное средство в полном соответствии с рекомендациями лечащего врача. При появлении сомнений посоветуйтесь с лечащим врачом или работником аптеки.
Рекомендуемые дозы:
Болезнь Крона: 5 мк/кг в 0, 2, 6 недели, затем каждые 8 недель. Некоторым взрослым пациентам с первоначальным ответом на терапию, но с утратой ответа впоследствии, может помочь повышение дозы до 10 мг/кг.
Болезнь Крона у пациентов детского возраста: 5 мк/кг в 0, 2, 6 недели, затем каждые 8 недель.
Язвенный колит: 5 мк/кг в 0, 2, 6 недели, затем каждые 8 недель.
Язвенный колит у пациентов детского возраста: 5 мк/кг в 0, 2, 6 недели, затем каждые 8 недель.
Ревматоидный артрит: в комбинации с метотрексатом, 3 мг/кг в 0, 2, 6 недели, затем каждые 8 недель. Некоторым пациентам может помочь повышение дозы до 10 мг/кг или введение лекарственного средства с частотой каждые 4 недели.
Анкилозирующий спондилит: 5 мк/кг в 0, 2, 6 недели, затем каждые 6 недель.
Псориатический артрит и бляшковидный псориаз: 5 мк/кг в 0, 2, 6 недели, затем каждые 8 недель.
Как применять лекарственное средство Инфлимаб
Инфлимаб применяется путем медленного (на протяжении не менее 2 часов) внутривенного вливания индивидуальной дозы, рассчитанной с учетом массы тела пациента. Кратность введения и доза лекарственного средства определяется лечащим врачом в зависимости от конкретной клинической ситуации. Раствор для введения приготавливается непосредственно перед применением препарата.
Подобно всем лекарственным средствам, Инфлимаб может вызывать нежелательные реакции, хотя и не у каждого пациента.
Если Вы считаете, что у Вас возникли побочные реакции на проводимую терапию, немедленно сообщите об этом Вашему лечащему врачу.
Наиболее частые нежелательные реакции (>10%): инфекции (верхних дыхательных путей, придаточных пазух носа, фарингиты), реакции, связанные с инфузией, головная боль и боль в животе.
Сообщение о нежелательных реакциях
Информацию о нежелательных реакциях направляйте на электронный адрес: fv@nativita.com или проконсультируйтесь с Вашим лечащим врачом. Данная рекомендация распространяется на любые возможные нежелательные реакции, в том числе – на те, что не были перечислены в листке-вкладыше.
Сообщая о нежелательных реакциях, Вы помогаете нам получить больше сведений о безопасности лекарственного средства.
Не применяйте лекарственное средство Инфлимаб после истечения срока годности, указанного на картонной пачке или блистере после слов «годен до». Датой истечения срока годности является последний день соответствующего месяца.
Условия хранения и транспортировки
Хранить при температуре от 2 °C до 8 °C в защищенном от света месте. Не замораживать. Хранить в недоступном для детей месте. После хранения лекарственного средства при температуре от 15 °C до 25 °C повторное хранение в холодильнике не допускается.
Действующее вещество: инфликсимаб – 100 мг.
Вспомогательные вещества: натрия гидрофосфата дигидрат – 6,10 мг, натрия дигидрофосфата дигидрат – 2,20 мг, сахароза – 500 мг, полисорбат 80 – 0,50 мг.
Лиофилизат в виде плотной массы белого цвета без признаков расплавления, не содержащий посторонних включений.
Лекарственное средство помещают во флаконы бесцветного нейтрального стекла I гидролитического класса, укупоренные резиновыми пробками, с обкаткой алюминиевыми колпачками с пластиковой крышкой типа «flip-off». На каждый флакон наклеивают самоклеящуюся этикетку. По 1 флакону вместе с инструкцией по медицинскому применению помещают в пачку из картона.
Владелец регистрационного удостоверения:
СООО «НАТИВИТА», Республика Беларусь,
211361, Витебская область, г.п. Бешенковичи, ул. Строителей, 3
тел./факс: +375213163164
E-mail: info@nativita.com

Кинерет® (раствор для подкожного введения, 150 мг/мл), инструкция по медицинскому применению РУ № ЛП-006746
Дата последнего изменения: 15.03.2022
Особые отметки:
![]()
![]()
Содержание
- Действующее вещество
- ATX
- Фармакологическая группа
- Лекарственная форма
- Состав
- Характеристика
- Описание лекарственной формы
- Фармакокинетика
- Фармакодинамика
- Показания
- Противопоказания
- Применение при беременности и кормлении грудью
- Способ применения и дозы
- Побочные действия
- Взаимодействие
- Передозировка
- Особые указания
- Форма выпуска
- Условия отпуска из аптек
- Условия хранения
- Срок годности
- Отзывы
Действующее вещество
ATX
Фармакологическая группа
Лекарственная форма
Раствор
для подкожного введения
Состав
В
0,67 мл раствора в одном предварительно заполненном шприце содержится:
Действующее вещество:
Анакинра
— 100 мг
Вспомогательные вещества:
Лимонная
кислота, натрия хлорид, динатрия эдетата дигидрат, полисорбат-8,0, вода для
инъекций.
Характеристика
Анакинра
представляет собой рекомбинантную версию антагониста рецептора интерлейкина-1
(IL‑lra) человека, продуцированного штаммом Е. coli с использованием технологии рекомбинантной ДНК.
Описание лекарственной формы
Прозрачный
или опалесцирующий, от бесцветного до беловатого цвета раствор, который может
содержать аморфные полупрозрачные или белые частицы, относящиеся к препарату
Фармакокинетика
Абсолютная
биодоступность анакинры после подкожной болюсной инъекции в дозе 70 мг у
здоровых субъектов (n = 11) составляет 95%. Процесс абсорбции является
фактором, ограничивающим скорость выведения анакинры из плазмы после подкожной
инъекции.
У
пациентов на фоне лечения препаратом Кинерет® максимальные
концентрации анакинры в плазме обнаруживали спустя 3–7 час после
подкожного введения анакинры в клинически значимых дозах (от 1 до 2 мг/кг;
n = 18). Концентрация в плазме уменьшалась без очевидной фазы распределения, а
период полувыведения составлял от 4 до 6 час. У пациентов, получавших препарат
Кинерет®, не наблюдали непредвиденного накопления анакинры после
ежедневного подкожного введения в течение до 24 недель. Средние (SD) показатели
клиренса (CL/F) и объема распределения (Vd/F) по результатам популяционного
фармакокинетического анализа данных 35 пациентов составили 105 (27) мл/мин
и 18,5 (11) л, соответственно. Данные, полученные в исследованиях на людях
и животных, показали, что почки являются основным органом, ответственным за
выведение анакинры. Клиренс анакинры у пациентов, получавших препарат Кинерет®,
возрастал с увеличением клиренса креатинина (КК).
Влияние
демографических параметров на фармакокинетику анакинры оценивали с помощью
популяционного фармакокинетического анализа данных 341 пациента, получавшего
ежедневно подкожно анакинру в дозах 30, 75 и 150 мг в течение до 24
недель. Оцениваемый клиренс анакинры увеличивался с увеличением КК и массы
тела. Результаты популяционного фармакокинетического анализа показали, что
среднее значение плазменного клиренса после подкожной болюсной инъекции
приблизительно на 14% выше у мужчин, чем у женщин, и приблизительно на 10% выше
у лиц в возрасте <65 лет, чем у испытуемых лиц в возрасте 65 лет.
Тем не менее, после коррекции по КК, массе тела, полу и возрасту существенных
факторов, влияющих на показатель среднего плазменного клиренса, выявлено не
было. Коррекция дозы препарата не требуется с учетом возраста или пола.
У
пациентов с CAPS была отмечена приблизительная линейность дозы с незначительной
тенденцией к более высокому, чем пропорциональному ее увеличению.
Фармакокинетические данные у детей <4 лет недостаточны, но имеется
клинический опыт применения у детей в возрасте от 8 месяцев. При инициации
лечения в рекомендуемой суточной дозе 1–2 мг/кг проблем с безопасностью
применения препарата выявлено не было. Фармакокинетические данные отсутствуют в
пожилой возрастной группе пациентов с CAPS. Было продемонстрировано
распределение препарата в спинномозговой жидкости. Медиана равновесной
нормализованной по дозе концентрации анакинры у пациентов в возрасте
3–17 лет с системным ювенильным идиопатическим артритом (Systemic Juvenile
Idiopathic Arthritis, SJIA) в течение 28 недель была сопоставима с таковой у
пациентов, получающих препарат Кинерет® по другим показаниям.
Печеночная недостаточность
Исследование
было проведено у 12 пациентов с дисфункцией печени (класс В по классификации
Чайлд-Пью), получивших однократную дозу 1 мг/кг внутривенно.
Фармакокинетические параметры пациентов и здоровых добровольцев значительно не
различались, за исключением показателя клиренса, который у пациентов был ниже
примерно на 30%. Соответствующее снижение КК было отмечено у пациентов с
печеночной недостаточностью. Таким образом, снижение клиренса, наиболее
вероятно, объясняется снижением функции почек у данной популяции пациентов. Эти
данные подтверждают, что для пациентов с печеночной недостаточностью класса В
по классификации Чайлд‑Пью коррекция дозы не требуется. Для пациентов с
печеночной недостаточностью средней степени тяжести коррекция дозы не требуется
(класс В по классификации Чайлд-Пью). Пациентам с тяжелой печеночной
недостаточностью следует проводить терапию препаратом Кинерет® с
осторожностью.
Почечная недостаточность
Среднее
значение плазменного клиренса на фоне лечения препаратом Кинерет®
пациентов с почечной недостаточностью легкой (КК 50–80 мл/мин) и средней
(КК 30–49 мл/мин) степени тяжести было снижено на 16% и 50%,
соответственно. У пациентов с почечной недостаточностью тяжелой степени тяжести
и терминальной стадией (КК <30 мл/мин) среднее значение клиренса плазмы
было снижено на 70% и 75%, соответственно. Менее 2,5% от введенной дозы
препарата Кинерет® было выведено с использованием гемодиализа или
постоянного амбулаторного перитонеального диализа. Эти данные подтверждают, что
для пациентов с почечной недостаточностью легкой степени тяжести (КК
50–80 мл/мин) коррекция дозы не требуется.
Фармакодинамика
Анакинра
блокирует биологическую активность интерлейкина-1а (ИЛ-1а) и
интерлейкина-1β (ИЛ‑1β) путем конкурентного ингибирования их
связей с рецептором интерлейкина-1 типа I. Интерлейкин‑1 (ИЛ-1) является
ключевым провоспалительным цитокином, медиатором многих клеточных ответов,
включая те, которые имеют важное значение при синовиальном воспалении.
Анакинра
ингибирует реакции, индуцированные ИЛ-1 in
vitro, в том числе, индукцию оксида азота и простагландина Е: и/или
синтез коллагеназы синовиальными клетками, фибробластами и хондроцитами.
Спонтанные
мутации в гене CIAS1/NLRP3 были обнаружены у большинства пациентов с
криопирин-ассоциированными периодическими синдромами (CAPS). CIAS1/NLRP3
кодирует криопирин, компонент инфламмасомы. Активация инфламмасомы приводит к
протеолитической матурации и секреции ИЛ-1β, который вызывает широкий
спектр реакций, включая системное воспаление. У пациентов с CAPS, не получавших
лечение, отмечают повышенный уровень С-реактивного белка (СРБ), амилоида А
(САА) и ИЛ-6 в сыворотке крови по сравнению с нормальными уровнями. Было
отмечено, что назначение препарата Кинерет® приводит к снижению
уровней острофазных белков и уменьшению экспрессии ИЛ-6. Снижение уровней
острофазных белков было зарегистрировано в течение первых недель лечения.
У
пациентов с семейной средиземноморской лихорадкой (Family Mediterranian Fever,
FMF) мутация гена MEFV, кодирующего пирин, приводит к нарушению функции и
избыточной продукции интерлейкина-1β (ИЛ-1β) в инфламмасоме пациента
с FMF. У пациентов с FMF, не получавших лечения, наблюдается увеличение уровней
СРБ и САА. Применение препарата Кинерет® приводит к уменьшению
уровней острофазных белков (например, СРБ и САА).
Болезнь
Стилла, помимо артрита различной степени тяжести, характеризуется системными
воспалительными явлениями, такими как резкая лихорадка, кожная сыпь,
гепатоспленомегалия, серозит и повышение уровней острофазных белков,
обусловленными активностью ИЛ-1. Системное действие ИЛ-1 проявляется в виде
температурной реакции гипоталамуса и гипералгезии. Роль ИЛ-1 в патогенезе
болезни Стилла была продемонстрирована ex
vivo и в исследованиях экспрессии генов.
Показания
Синдромы периодической лихорадки
Препарат
Кинерет® показан взрослым, подросткам и детям в возрасте от 8 месяцев
и старше с массой тела от 10 кг и выше для лечения следующих
аутовоспалительных синдромов периодической лихорадки:
Криопирин-ассоциированных периодических синдромов (CAPS),
включая:
—
Мультисистемное
воспалительное заболевание неонатального возраста (NOMID)/хронический
младенческий неврологический кожно-артикулярный синдром (CINCA)
—
Синдром
Макла-Уэллса (MWS)
—
Семейный
Холодовой аутовоспалительный синдром (FCAS)
Семейной средиземноморской лихорадки (FMF)
Пациентам
с FMF препарат Кинерет® следует назначать, по возможности, совместно
с колхицином.
Болезнь Стилла
Препарат
Кинеретк показан взрослым, подросткам и детям в возрасте от 8 месяцев и старше
с массой тела от 10 кг и выше для лечения, включая системный ювенильный
идиопатический артрит (SJIA) и болезнь Стилла, диагностированную у взрослых
(AOSD), с системными симптомами умеренной и высокой степени активности
заболевания, или у пациентов с длительной активностью заболевания после лечения
нестероидными противовоспалительными препаратами (НПВП) или глюкокортикостероидами.
Препарат
Кинерет® можно назначать в виде монотерапии или в комбинации с
другими противовоспалительными препаратами и базисными противоревматическими
препаратами (БПРП).
Противопоказания
Повышенная
чувствительность к действующему веществу или любым вспомогательным веществам
или производным белка Е. coli.
Препарат
Кинерет® не следует назначать пациентам с нейтропенией (абсолютное
число нейтрофилов, ANC <1,5 × 109/л).
С осторожностью
—
У пациентов,
имеющих в анамнезе рецидивирующие инфекции или предрасполагающие факторы,
которые могут привести к развитию инфекции; повышение активности печеночных
ферментов в анамнезе или печеночную недостаточность тяжелой степени тяжести;
почечную недостаточность средней степени тяжести (КК 30–59 мл/мин);
бронхиальную астму; злокачественные новообразования, в том числе, в анамнезе
(см. раздел «Особые указания»);
—
У пациентов,
которым требуется вакцинация (см. раздел «Особые указания»);
—
У пожилых
пациентов (см. раздел «Особые указания»);
—
У пациентов,
принимающих одновременно варфарин или фенитоин (см. раздел «Взаимодействие с
другими лекарственными средствами»).
Применение при беременности и кормлении грудью
Беременность
Данные
о применении анакинры у беременных женщин ограничены. Результаты исследований
на животных не указывают на наличие прямой или косвенной репродуктивной
токсичности.
В
качестве меры предосторожности рекомендуется избегать применения анакинры во
время беременности, а также женщинам детородного возраста, не использующим
контрацепцию.
Грудное вскармливание
Неизвестно,
проникает ли анакинра/метаболиты в грудное молоко. Риск для новорожденного
ребенка/младенца не может быть исключен. Во время лечения препаратом Кинерет®
грудное вскармливание следует прекратить.
Способ применения и дозы
Раствор
для подкожного введения.
Лечение
препаратом Кинеретк должно быть назначено врачами-специалистами, имеющими опыт
в диагностике и лечении CAPS, FMF и болезни Стилла, и проводиться под их
контролем.
Препарат
Кинерет® вводится путем подкожной инъекции. Препарат Кинерет®
поставляется готовым к применению в градуированном предварительно заполненном
шприце. Градуированный предварительно заполненный шприц предназначен для
дозирования от 20 до 100 мг анакинры. Поскольку минимальная доза в шприце
составляет 20 мг, он не подходит для лечения детей с массой тела менее
10 кг.
Во
избежание чувства дискомфорта в месте инъекции рекомендуется чередовать места
инъекции. Для облегчения симптомов реакций в месте инъекции рекомендуется
охлаждение места инъекции, нагревание раствора препарата до комнатной
температуры, наложение на место инъекции холодных пакетов (до ее проведения и
после), применение кортикостероидов и антигистаминных препаратов для местного
применения.
CAPS: Взрослые, подростки и дети в возрасте от 8 месяцев и
старше и массой тела от 10 кг и выше
Начальная доза:
Рекомендуемая
начальная доза при всех подтипах CAPS составляет 1–2 мг/кг/день в виде
подкожной инъекции. Терапевтический ответ проявляется, главным образом, в виде
уменьшения клинических симптомов, таких как лихорадка, сыпь, боли в суставах и
головная боль, а также снижением уровней острофазных белков (СРБ/САА) или
частоты обострений.
Поддерживающая доза при CAPS легкой степени тяжести (FCAS, MWS
легкой степени тяжести):
Как
правило, клинические и лабораторные симптомы успешно контролируются путем
строгого соблюдения рекомендуемой начальной дозы (1–2 мг/кг/день).
Поддерживающая доза при CAPS тяжелой степени тяжести (MWS и
NOM1D/CINCA):
В
течение 1–2 месяцев может потребоваться увеличение дозы с учетом
терапевтического ответа. Обычная поддерживающая доза при CAPS тяжелого течения
составляет 3–4 мг/кг/день, и она может быть увеличена до максимальной дозы
8 мг/кг/день.
В
дополнение к оценке клинических симптомов и воспалительных маркеров при CAPS
тяжелого течения, рекомендуется проводить оценку воспаления центральной нервной
системы (ЦНС), в том числе внутреннего уха (с помощью магниторезонансной
томографии (МРТ) или компьютерной томографии (КТ), люмбальной пункции и
аудиологического исследования) и органов зрения (офтальмологическая оценка)
после первоначального 3‑месячного курса лечения, а затем каждые 6
месяцев, до тех пор, пока не будет определен эффективный режим терапии. Когда
контроль состояния пациента успешно достигнут, мониторинг клинических,
офтальмологических показателей и параметров ЦНС можно проводить ежегодно.
FMF:
Рекомендуемая
доза для пациентов с массой тела 50 кг и выше составляет 100 мг/день
в виде подкожной инъекции. Пациентам с массой тела менее 50 кг следует
рассчитать дозу с учетом массы тела и рекомендуемой дозы 1–2 мг/кг/день.
Болезнь Стилла:
Рекомендуемая
доза для пациентов с массой тела от 50 кг и более составляет
100 мг/день в виде подкожной инъекции. Пациентам с массой тела менее
50 кг следует рассчитать дозу с учетом массы тела и начальной дозы
1–2 мг/кг/день.
Ответ
на лечение следует оценить через 1 месяц: в случае стойких системных проявлений
доза у детей может быть скорректирована или целесообразность продолжения терапии
препаратом Кинерет® должна быть повторно оценена лечащим врачом.
Применение у пожилых пациентов (≥65 лет)
CAPS: данные о
применении препарата у пожилых пациентов с CAPS ограничены. Предположительно,
коррекция дозы препарата не требуется.
Болезнь Стилла:
данные о применении препарата у пожилых пациентов ограничены. Предположительно,
коррекция дозы препарата не требуется.
Применение у детей (в возрасте <18 лет)
Данные
о применении препарата у детей в возрасте до 8 месяцев отсутствуют.
CAPS: режим дозирования
и способ применения у детей в возрасте 8 месяцев и старше с массой тела
10 кг и выше соответствует режиму применения у взрослых пациентов с CAPS и
зависит от массы тела.
FMF: детям с массой
тела менее 50 кг дозу препарата рассчитывают с учетом массы тела и
рекомендуемой дозы 1–2 мг/кг/день; пациентам с массой тела от 50 кг и
более препарат следует вводить в дозе 100 мг/день. У детей с недостаточным
ответом на терапию дозу препарата можно повысить до 4 мг/кг/день.
Данные
об эффективности препарата Кинерет® у детей с FMF в возрасте младше
2 лет ограничены.
Болезнь Стилла:
детям с массой тела менее 50 кг дозу препарата рассчитывают с учетом массы
тела и начальной дозы 1–2 мг/кг/день; пациентам с массой тела от
50 кг и более препарат следует вводить в дозе 100 мг/день. У детей с
недостаточным ответом на терапию дозу препарата можно повысить до
4 мг/кг/день.
Применение у пациентов с нарушением функции печени
Для
пациентов с печеночной недостаточностью средней степени тяжести (класс В по
классификации Чайлд-Пью) коррекция дозы не требуется. Пациентам с тяжелой
печеночной недостаточностью (класс С по классификации Чайлд-Пью) следует
проводить терапию препаратом Кинерет® с осторожностью.
Применение у пациентов с нарушением функции почек
Для
пациентов с почечной недостаточностью легкой степени тяжести (КК
60–89 мл/мин) коррекция дозы не требуется. Препарат Кинерет®
следует применять с осторожностью у пациентов с почечной недостаточностью
средней степени тяжести (КК 30–59 мл/мин). У пациентов с почечной
недостаточностью тяжелой степени тяжести (КК <30 мл/мин) и терминальной
стадией почечной недостаточности, включая пациентов на диализе, следует
предусмотреть введение назначенной дозы препарата Кинерет® через
день.
Если препарат Кинерет® введен в большей дозе, чем
это было необходимо
При
случайном введении препарата Кинерет® в дозе, превышающей
необходимую, не должно возникнуть серьезных проблем. Тем не менее, в подобном
случае пациенту следует обратиться к врачу. В любом случае, если пациент
почувствовал себя нехорошо, он/она должен/должна незамедлительно обратиться к
врачу.
Если пропущено введение дозы препарата Кинерет®
Если
пропущено введение дозы препарата Кинерет®, пациенту следует
обратиться к врачу, чтобы обсудить, когда следует вводить следующую дозу.
Инструкция по подготовке и выполнению инъекции препарата
Кинерет®
Данный
раздел содержит информацию о том, как сделать себе или своему ребенку инъекцию
препарата Кинерет®. Не следует вводить препарат Кинерет®
себе или своему ребенку, если Вы не прошли инструктаж у врача или медицинской
сестры. Если у вас есть вопросы о том, как вводить препарат, пожалуйста,
обратитесь за помощью к врачу или медицинской сестре.
Препарат
Кинерет® представляет собой стерильный раствор, не содержащий
консервантов. Только для однократного применения.
Шприц
не следует встряхивать. Перед инъекцией необходимо дать нагреться шприцу до
комнатной температуры.
Перед
введением нужно проверить внешний вид раствора на наличие механических включений
и изменения цвета. Вводить можно только прозрачный или слабо опалесцирующий, от
бесцветного до беловатого цвета раствор, который может содержать аморфные
полупрозрачные или белые частицы, относящиеся к препарату.
Присутствие
таких частиц не влияет на качество препарата.
Шприц
предназначен только для одноразового применения. Любые неиспользованные остатки
препарата следует утилизировать.
Как сделать инъекцию самостоятельно или с помощью другого
лица с использованием препарата Кинерет® в градуированном
предварительно заполненном шприце
Вводить
препарат себе или своему ребенку необходимо в одно и то же время каждый день.
Поскольку препарат Кинерет® предназначен для подкожных инъекций, его
вводят только под кожу.
Для
выполнения подкожной инъекции препарата себе или своему ребенку вам
понадобятся:
—
Препарат Кинерет®
в градуированном предварительно заполненном шприце;
—
Спиртовые
салфетки или аналоги;
—
Стерильная марля
или ткань.
Что вам следует сделать перед подкожным введением препарата
Кинерет® себе или своему ребенку?
1. Достаньте
препарат Кинерет® в градуированном предварительно заполненном шприце
из холодильника.
2. Не
встряхивайте шприц.
3. Проверьте
дату истечения срока годности на этикетке предварительно заполненного шприца (в
редакции «До:»). Не используйте препарат Кинерет® после истечения
срока годности, указанного на упаковке.
4. Проверьте
внешний вид раствора препарата Кинерет® в шприце. Раствор должен
быть прозрачным или слабо опалесцирующим, от бесцветного до беловатого цвета.
Раствор может содержать аморфные полупрозрачные или белые частицы, относящиеся
к препарату. Присутствие этих частиц не влияет на качество препарата. Раствор
не следует вводить, если он изменил цвет или стал непрозрачным, или если
имеются какие-либо другие частицы, отличные от прозрачно-белых частиц.
5. Для
облегчения симптомов реакций в месте инъекции препарата оставьте шприц при
комнатной температуре в течение приблизительно 30 минут или осторожно подержите
его в руке в течение нескольких минут. Не нагревайте Кинерет® каким-либо
иным способом (например, не нагревайте его в микроволновой печи или при помощи
горячей воды).
6. Не
снимайте защитный колпачок со шприца, если Вы не готовы вводить препарат.
7. Тщательно
вымойте руки.
8. Найдите
удобную, хорошо освещенную и чистую поверхность и поместите все необходимое
оборудование перед собой.
9. Убедитесь,
что Вы знаете, какую дозу препарата Кинеретк назначил ваш врач; от 20 до
90 мг, 100 мг или выше.
—
Если врач
назначил дозу 100 мг, смотрите раздел «Как приготовить дозу 100 мг».
—
Если врач
назначил более низкую дозу, смотрите раздел «Как приготовить дозу от 20 до
90 мг».
Как приготовить дозу 100 мг
Перед
введением препарата Кинерет® вам следует выполнить следующее:
1. Возьмите
шприц и осторожно снимите с иглы защитный колпачок, не вращая его. Потяните
колпачок прямо, как показано на Рис. А. Не прикасайтесь к игле и не нажимайте
на поршень. Сразу выбросьте колпачок от иглы.
Рисунок
А
2. Вы
можете заметить наличие небольшого пузырька воздуха в шприце. Вам не следует
удалять пузырьки воздуха перед инъекцией. Введение раствора с пузырьком воздуха
безопасно.
3. Теперь
Вы можете использовать предварительно заполненный шприц согласно описанию в
следующих разделах «Куда следует делать себе инъекцию?» и «Как сделать себе
инъекцию?».
Как приготовить дозу от 20 до 90 мг
Перед
введением препарата Кинерет® вам следует выполнить следующее:
1.
Возьмите шприц и осторожно снимите с иглы колпачок, не вращая его. Потяните
колпачок прямо, как показано на Рис. А. Не прикасайтесь к игле и не нажимайте
на поршень. Сразу выбросьте колпачок от иглы.
2.
Вам следует держать шприц в одной руке, направляя иглу прямо вверх, как
показано на Рис. Б. Поместите и держите большой палец на штоке поршня до тех
пор, пока на кончике иглы не появится крошечная капля жидкости.
Рисунок
Б
3.
Переверните шприц таким образом, чтобы игла теперь была направлена вниз.
Поместите стерильную марлю или ткань на плоскую поверхность и держите над ней
шприц с иглой, направленной в сторону марли или ткани, как показано на Рис. В.
Убедитесь в том, что игла не касается марли или ткани.
4.
Поместите большой палец на шток поршня и нажимайте на него до тех пор, пока
передняя часть поршня не дойдет до метки на шкале, указывающей на необходимую
вам дозу препарата Кинерет8 (ваш врач проинформирует вас о назначенной дозе
препарата). Выпущенная струя жидкости впитается марлей или тканью, как показано
на Рис. В.
5.
Если вы не смогли установить правильную дозу, утилизируйте шприц и используйте
новый.
6.
Теперь вы можете использовать предварительно заполненный шприц согласно
описанию в следующих разделах «Куда следует делать себе инъекцию?» и «Как
сделать себе инъекцию?».
Куда следует делать себе инъекцию?
Наиболее
подходящие участки для выполнения инъекции себе или своему ребенку (смотрите
Рис. Г):
—
живот (за
исключением области вокруг пупка)
—
верхняя часть
бедра
—
верхние наружные
области ягодиц;
—
наружная область
плеча
Рисунок
Г
Каждый
раз меняйте место инъекции, чтобы избежать развития болезненности в одной
области. При введении препарата с посторонней помощью может быть также
задействована задняя поверхность вашего плеча.
Как сделать себе инъекцию?
1.
Продезинфицируйте кожу с помощью спиртовых салфеток и зажмите кожу между
большим и указательным пальцами, не сдавливая ее.
2.
Введите иглу в кожу полностью, как было вам показано медицинской сестрой или
врачом.
3.
Вводите раствор препарата медленно и равномерно в образовавшуюся кожную
складку, как показано на Рис. Д.
4.
После введения препарата, извлеките иглу и отпустите кожу.
5.
Любые неиспользованные остатки препарата должны быть утилизированы. Используйте
один шприц только для одной инъекции. Не используйте шприц повторно, поскольку
это может стать причиной развития инфекции.
Запомните
Если
у вас есть какие-либо проблемы, пожалуйста, незамедлительно обращайтесь к
своему врачу или медицинской сестре за помощью и советом.
Утилизация использованных шприцев и материалов
—
Не надевайте
колпачок на использованные иглы
—
Держите
использованные шприцы в недоступном для детей месте
—
Никогда не
кладите использованный шприц в обычное мусорное ведро с бытовыми отходами
—
Если вам нужна
доза препарата менее 100 мг, как было сказано выше, следует удалить
избыток препарата из шприца на марлю или ткань. После инъекции утилизируйте
мокрую марлю или ткань с вашим шприцем и очистите поверхность чистой тканью
—
Использованные
шприцы и марлю или ткань следует утилизировать согласно местным законодательным
актам. Данные меры помогут защитить окружающую среду.
Побочные действия
Краткая характеристика профиля безопасности
Данные
о нежелательных лекарственных реакциях (НЛР) у пациентов с CAPS получены в
открытом КИ у 43 пациентов с NOMID/CINCA на фоне терапии препаратом Кинерет®,
длительностью до 5 лет и показателем общей экспозиции 159,8 пациенто-лет.
В ходе 5‑летнего КИ у 14 пациентов (32,6%) было зарегистрировано 24
серьезных нежелательных явления (СНЯ). Одиннадцать СНЯ у 4 пациентов (9,3%)
были расценены как связанные с применением препарата Кинерет®. Ни у
одного пациента лечение препаратом Кинерет® не было прекращено из-за
HЛP.
Данные
о нежелательных явлениях (НЯ) у пациентов с болезнью Стилла основаны частично
на результатах открытого и на результатах слепого плацебо-контролируемого
исследования 15 пациентов с SJIA, получавших терапию длительностью до
1,5 лет, а также результатах рандомизированного двойного слепого,
плацебо-контролируемого исследования с участием 12 пациентов взрослого и
детского возраста с болезнью Стилла (6 пациентов в группе препарата Кинерет®
и 6 пациентов в группе плацебо), получавших терапию длительностью 12 недель с
дополнительным продлением на 4 недели.
Кроме
того, результаты долгосрочных неинтервенционных исследований безопасности на
306 пациентах с болезнью Стилла, пострегистрационные отчеты о НЯ и
опубликованные исследования подтверждают эти данные.
Данные
о НЯ у пациентов с FMF основаны на пострегистрационных сообщениях о НЯ и
результатах опубликованных исследований.
В
этих исследованиях и в пострегистрационных сообщениях о НЛР отсутствуют данные
о том, что общий профиль безопасности у пациентов с CAPS, FMF или болезнью
Стилла отличается от такового у пациентов, у которых препарат Кинерет®
применяли по другим показаниям, за исключением увеличения частоты НЯ со стороны
печени у пациентов с болезнью Стилла согласно пострегистрационным наблюдениям.
Таким образом, приведенная ниже таблица НЛР применима к препарату Кинерет®
при лечении CAPS, FMF и болезни Стилла. При долгосрочной терапии CAPS и болезни
Стилла профиль безопасности не изменяется.
Ниже
представлены НЛР, которые классифицированы в соответствии с поражением органов
и систем органов (медицинский словарь для нормативно-правовой деятельности
MedDRA) и частотой возникновения.
|
Класс |
Частота |
Нежелательная |
|
Инфекционные |
Часто |
Тяжелые |
|
Нарушения |
Часто |
Нейтропения Тромбоцитопения |
|
Нарушения |
Нечасто |
Аллергические |
|
Нарушения |
Очень |
Головная |
|
Нарушения |
Нечасто |
Повышение |
|
Частота |
Неинфекционный |
|
|
Нарушения |
Нечасто |
Сыпь |
|
Общие |
Очень |
Реакции |
|
Лабораторные |
Очень |
Повышение |
Если
любые из указанных в инструкции побочных эффектов усугубляются, или пациент
заметил любые другие побочные эффекты, не указанные в инструкции, следует
сообщить об этом врачу.
Тяжелые инфекции
В
КИ у 43 пациентов с CAPS за 5-летний период наблюдения частота тяжелых инфекций
составляла 0,1/год, из которых чаще всего встречалась пневмония и
гастроэнтерит. Применение препарата Кинерет® было временно прекращено
у одного пациента; остальные пациенты продолжили лечение препаратом Кинерет®
во время течения инфекций.
Во
время наблюдения за 15 пациентами с SJIA длительностью 1,5 года, у одного
пациента развился тяжелый гепатит на фоне цитомегаловирусной инфекции. В
исследовании с участием 11 пациентов с болезнью Стилла (SJIA и AOSD),
рандомизированных в группы препарата Кинерет® (6 пациентов) и
плацебо (5 пациентов) и наблюдаемых на протяжении 16 недель, не сообщалось о
развитии серьёзных инфекций. В долгосрочном неинтервенционном исследовании
безопасности препарата Кинерет® с участием 306 пациентов детского
возраста с болезнью Стилла на протяжении более 9 лет (средняя продолжительность
курса терапии препаратом Кинерет® составила 17,0 (стандартное
отклонение 21,1) месяцев; медиана продолжительности терапии составила 8,9
месяцев) серьезные инфекции наблюдались у 13 пациентов. Данные
пострегистрационного отчета о НЯ и результаты опубликованных исследований не
выявили отличий в типе и степени тяжести инфекций у пациентов с FMF и пациентов
с CAPS или болезнью Стилла.
В
ходе КИ и пострегистрационного применения препарата были отмечены редкие случаи
инфекций условно-патогенными организмами, которые включали грибковые,
микобактериальные,
бактериальные и вирусные патогены. Инфекции были отмечены во всех системах
органов и были зарегистрированы у пациентов, получавших препарат Кинерет®
отдельно или в сочетании с иммунодепрессантами.
Нейтропения
В
плацебо-контролируемых КИ препарата Кинерет® по показаниям, отличных
от CAPS, FMF и болезни Стилла, в ходе лечения было отмечено незначительное
уменьшение средних значений общего числа лейкоцитов и абсолютного числа
нейтрофилов. Нейтропения (ANC <1,5 ? 109/л) была
зарегистрирована у 2,4% пациентов, получавших препарат Кинерет® по
сравнению с 0,4% пациентов, получавших плацебо. Ни у одного из этих пациентов
не было диагностировано острых инфекций, связанных с нейтропенией. Из 43
пациентов с CAPS, наблюдавшихся в течение пяти лет в рамках КИ, нейтропения
была отмечена у 2 пациентов. В обоих случаях нейтропения разрешилась в ходе
продолжения лечения препаратом Кинерет®.
За
полтора года наблюдения за 15 пациентами с SJIA у одного пациента был отмечен
один эпизод транзиторной нейтропении. В исследовании с участием 11 пациентов с
болезнью Стилла (SJIA и AOSD), рандомизированных в группы препарата Кинерет®
(6 пациентов) и плацебо (5 пациентов) и наблюдаемых на протяжении 16 недель, не
сообщалось о развитии нейтропении. В долгосрочном неинтервенционном
исследовании безопасности препарата Кинерет® с участием 306
пациентов детского возраста с болезнью Стилла на протяжении более 9 лет
(средняя продолжительность курса терапии препаратом Кинерет®
составила 17,0 (стандартное отклонение 21,1) месяцев; медиана продолжительности
терапии составила 8,9 месяцев) сообщалось о 5 случаях нейтропении, включая 1
случай фебрильной нейтропении.
Тромбоцитопения
В
ходе КИ препарата Кинерет®, назначаемого по показаниям, не
включающим CAPS, FMF и болезнь Стилла, тромбоцитопения была зарегистрирована у
1,9% пациентов в группе препарата Кинерет® по сравнению с 0,3%
пациентов в группе плацебо. Тромбоцитопения была легкой (количество тромбоцитов
>75 ? 109/л). У пациентов с CAPS также наблюдали легкую
тромбоцитопению. В ходе пострегистрационного применения препарата Кинерет®
была зарегистрирована тромбоцитопения, в том числе, эпизодически отмечали
случаи тяжелой тромбоцитопении (количество тромбоцитов
<10 ? 109/л).
Злокачественные опухоли
В
рамках КИ препарата Кинерет®, назначаемого по показаниям, не
включающим CAPS, FMF и болезнь Стилла, общая частота случаев злокачественных
опухолей была одинаковой в группе пациентов, принимавших препарат Кинерет®,
и в группе, принимавшей плацебо, и не отличалась от частоты случаев,
зарегистрированных в целом в исследуемых популяциях. Кроме того, общая частота
случаев злокачественных опухолей не увеличивалась на протяжении трех лет
применения препарата Кинерет®.
Синдром активации макрофагов (Macrophage
activation syndrome, MAS)
В
ходе пострегистрационного применения препарата были получены сообщения о
развитии MAS у пациентов с болезнью Стилла, получавших лечение препаратом
Кинерет®. Пациенты с болезнью Стилла имеют повышенный риск
спонтанного развития MAS. Причинно-следственная связь между терапией препаратом
Кинеретом8 и развитием MAS не установлена.
Аллергические реакции
Аллергические
реакции на препарат Кинерет8, включая анафилактические реакции,
ангионевротический отек, крапивницу, сыпь и зуд, встречались нечасто.
Большинство из этих реакций были представлены макулопапулезной или уртикарной
сыпью.
У
43 пациентов с CAPS, наблюдавшихся в течение пяти лет в рамках КИ, не было
серьезных аллергических реакций и случаев, требующих досрочного прекращения
лечения препаратом Кинерет®.
Во
время полуторагодичного наблюдения за 15 пациентами с SJIA не зарегистрировано
НЯ или требующих отмены терапии препаратом Кинерет® аллергических
реакций. В исследовании с участием 11 пациентов с болезнью Стилла (SJIA и
AOSD), рандомизированных в группы препарата Кинерет® (6 пациентов) и
плацебо (5 пациентов) и наблюдаемых на протяжении 16 недель, не сообщалось о
развитии аллергических реакций. В опубликованном рандомизированном
контролируемом исследовании у 12 пациентов с FMF, получавших Кинерет8 в течение
4 месяцев, не было зарегистрировано ни одной серьезной аллергической реакции, и
ни одно НЯ не потребовало отмены терапии препаратом Кинерет®.
Иммуногенность
В
ходе КИ у большинства пациентов с CAPS были обнаружены антитела к анакинре. Не
было отмечено клинически значимого влияния наличия антител к анакинре на
фармакокинетику, эффективность и безопасность препарата Кинерет®.
Кроме того, в КИ у 6% из 86 детей с JIA, по крайней мере, один раз за время
исследования обнаружены нейтрализующие антитела к препарату анакинра, при этом,
антитела не были выявлены среди 15 пациентов с подтипом SJIA. В КИ с участием 6
пациентов с болезнью Стилла (SJIA и AOSD), рандомизированных в группу анакинры
и получавших терапию на протяжении 12 недель, в какой-то период терапии были
обнаружены антитела к препарату у каждого пациента, но ни у одного из них не
было обнаружено нейтрализующих антител к анакинре.
Изменения функции печени
В
ходе КИ наблюдали случаи транзиторного повышения активности печеночных
ферментов. Это повышение не было связано с признаками или симптомами
повреждения клеток печени, за исключением одного пациента с SJIA, у которого
развился тяжелый гепатит, ассоциированный с цитомегаловирусной инфекцией. В
ходе пострегистрационного применения препарата Кинерет® были
получены сообщения о единичных случаях неинфекционного гепатита.
В
пострегистрационном периоде изменения функции печени были отмечены
преимущественно у пациентов с болезнью Стилла и у пациентов с
предрасполагающими факторами в анамнезе, например, имевших повышенную
активность трансаминаз до назначения препарата Кинерет®.
Реакции в месте инъекции
Реакции
в месте инъекции обычно возникают в течение 2 недель с момента введения
препарата, и исчезают в течение 4–6 недель. У пациентов, у которых ранее не
регистрировали реакций в месте инъекции, эти реакции редко развивались после
первого месяца терапии.
В
ходе КИ препарата Кинерет®, назначаемого по показаниям, отличным от
CAPS, FMF и болезни Стилла, наиболее частыми НЛР являлись реакции в месте
инъекции. Большинство (95%) реакций в месте инъекции были от легкой до
умеренной степени тяжести. Реакции в месте инъекции обычно характеризовались
одним или несколькими признаками, такими как: эритема, экхимоз, воспаление и
боль.
Из
43 пациентов с CAPS, наблюдавшихся в течение пяти лет в рамках КИ, ни один
пациент не прекратил лечение препаратом Кинерет® и не приостановил
его временно в связи с реакциями в месте инъекции.
Во
время полуторагодичного наблюдения за 15 пациентами с SJIA наиболее частыми
НЛР, ассоциируемыми с применением препарата Кинерет®, были реакции в
месте инъекции. Один из 15 пациентов прекратил терапию в связи с развитием
реакции в месте инъекции.
В
исследовании с участием 11 пациентов с болезнью Стилла (SJIA и AOSD),
рандомизированных в группы препарата Кинерет® (6 пациентов) и
плацебо (5 пациентов) и наблюдаемых на протяжении 12 недель, реакции в месте
инъекции средней степени тяжести развились в обеих группах лечения. Ни один из
пациентов не был исключен из исследования из-за этих реакций. В долгосрочном
неинтервенционном исследовании безопасности с участием 306 пациентов детского
возраста с болезнью Стилла на протяжении более 9 лет (средняя
продолжительность курса терапии препаратом Кинерет® составила 17,0
(стандартное отклонение 21,1) месяцев; медиана продолжительности терапии
составила 8,9 месяцев) частота развития реакций в месте инъекции средней или
тяжелой степени тяжести составила 1,6/100 пациенто-лет.
У
пациентов с FMF тип и частота реакций в месте инъекции были аналогичны таковым
при лечении других заболеваний. Случаи отмены терапии в связи с развитием
реакций в месте инъекции наблюдались также у пациентов с FMF.
Повышение холестерина в крови
У
775 пациентов, которые в рамках КИ по отличным от CAPS, FMF и болезни Стилла
показаниям получали препарат Кинерет® в суточных дозах 30 мг,
75 мг, 150 мг, 1 мг/кг или 2 мг/кг, наблюдали увеличение
общего содержания холестерина от 2,4% до 5,3% через 2 недели после начала
лечения препаратом Кинерет® вне зависимости от дозы препарата.
Подобные изменения показателей общего холестерина отмечали и через 24 недели
лечения препаратом Кинерет®. Лечение плацебо (n = 213)
сопровождалось уменьшением общего содержания холестерина приблизительно на 2,2%
на неделе 2 и на 2,3% на неделе 24. Данных о липопротеинах низкой плотности и
липопротеинах высокой плотности не имеется.
Применение у детей
Препарат
Кинерет® изучали у пациентов в возрасте от 8 месяцев до
<18 лет в течение 5 лет
у 36 пациентов с CAPS, у 21 пациента с SJIA и 71 пациента
с другими вариантами JIA. За исключением инфекций и связанных с ними симптомов,
которые чаще отмечались у пациентов в возрасте <2 лет, профиль
безопасности был сходным у детей всех возрастных групп. Помимо этого, в
долгосрочном неинтервенционном исследовании безопасности более 9 лет проводили
наблюдение за 306 пациентами детского возраста с болезнью Стилла.
Педиатрическая
и взрослая популяции пациентов имели сходный профиль безопасности; новых
клинически значимых НЛР отмечено не было.
Взаимодействие
Взаимодействие
между препаратом Кинерет® и другими лекарственными средствами
направленно не изучали. В рамках КИ не было отмечено взаимодействия между
препаратом Кинерет® и другими лекарственными средствами (в том числе
нестероидными противовоспалительными препаратами, глюкокортикостероидами и
базисными противоревматическими препаратами (БПРП)).
Одновременное применение препарата Кинерет® и
антагониста фактора некроза опухоли‑альфа (ФНО-α)
В
ходе КИ у пациентов (отличных от CAPS, FMF, болезни Стилла) на фоне основной
терапии метотрексатом, которые одновременно получали препарат Кинерет®
и этанерцепт, частота развития острых инфекций (7%) и нейтропении была выше,
чем у пациентов, получавших только этанерцепт или только препарат Кинерет®.
Одновременный прием препарата Кинерет® и этанерцепта или другого
антагониста фактора некроза опухоли-альфа (ФНО-α) не рекомендуется.
Субстраты цитохрома Р450
При
хроническом воспалении образование ферментов CYP450 подавляется
вследствие повышения уровня цитокинов (например, ИЛ-1). Таким образом, можно
ожидать, что для антагониста рецептора ИЛ-1, такого как анакинра, образование
ферментов CYP450 может быть нормализовано в ходе лечения. Это может
иметь клиническое значение для субстратов CYP450 с узким
терапевтическим индексом (например, варфарина и фенитоина). У пациентов,
применяющих препараты данного типа, в начале или конце лечения препаратом
Кинерет® целесообразно проведение оценки их терапевтического
эффекта, а также лабораторных показателей или концентрации этих препаратов с
последующей возможной коррекцией их дозы.
Передозировка
В
ходе КИ не было отмечено ограничивающей дозу токсичности. В рамках КИ по
отличным от CAPS показаниям 1015 пациентов получали препарат Кинерет®
внутривенно в дозах до 2 мг/кг/час на протяжении 72 часов. Профиль
нежелательных явлений в этих КИ не отличался от профиля безопасности в КИ, где
применяли более низкие дозы препарата Кинерет®.
Особые указания
Отслеживаемость
Для
улучшения отслеживаемое биологических лекарственных препаратов следует точно
записывать название и номер партии вводимого препарата.
Аллергические реакции
Аллергические
реакции, включая анафилактические реакции и ангионевротический отек,
встречались нечасто. Чаще всего отмечали макулопапулезную или уртикарную сыпь.
При появлении признаков тяжелой аллергической реакции введение препарата
Кинерет® следует прекратить и назначить надлежащее лечение.
Нежелательные явления со стороны печени
В
ходе КИ были отмечены случаи временного повышения активности печеночных
ферментов. Эти повышения не были связаны с признаками или симптомами
повреждения клеток печени, за исключением одного пациента с SJIA, у которого
развился тяжелый гепатит, ассоциированный с цитомегаловирусной инфекцией.
В
ходе пострегистрационного применения препарата сообщалось о НЯ со стороны
печени, не оказывавших влияния на ее функцию. Большинство этих пациентов
получали лечение по поводу болезни Стилла или имели предрасполагающие факторы в
анамнезе, например, повышенную активность трансаминаз. Кроме того, на фоне
терапии препаратом Кинерет® у пациентов с болезнью Стилла были
отмечены случаи неинфекционного гепатита, включая эпизодические случаи острой
печеночной недостаточности. Нарушения со стороны печени у пациентов с болезнью
Стилла преимущественно возникают в течение первого месяца лечения препаратом
Кинерет®. Следует рассмотреть вопрос о регулярной оценке активности
печеночных ферментов в течение первого месяца терапии, особенно у пациентов с
предрасполагающими факторами или при наличии симптомов, указывающих на
нарушение функции печени.
Оценку
эффективности и безопасности применения препарата Кинерет® у
пациентов с активностью аспартатаминотрансферазы/аланинаминотрансферазы
≥1,5 раза выше верхней границы нормы не проводили.
Тяжелые инфекции
В
ходе КИ препарата Кинерет® при показаниях, отличных от CAPS, FMF и
болезни Стилла, была отмечена взаимосвязь применения препарата Кинерет®
с увеличением заболеваемости тяжелыми инфекциями (1,8%) по сравнению с плацебо
(0,7%). Для небольшого числа пациентов с бронхиальной астмой, заболеваемость
острыми инфекциями была выше у пациентов, получавших препарат Кинерет®
(4,5%), по сравнению с пациентами, получавшими плацебо (0%); в основном были
диагностированы инфекции респираторного тракта. Оценку безопасности и
эффективности применения препарата Кинерет® у пациентов с
хроническими и острыми инфекциями не проводили.
Препарат
Кинерет® не следует назначать пациентам с активными инфекциями. Для
пациентов с CAPS или FMF существует риск обострения заболевания после
прекращения терапии препаратом Кинерет®. При условии тщательного
контроля лечение препаратом Кинерет® может быть продолжено и во
время тяжелой инфекции.
Синдром
активации макрофагов (MAS) — это известное, опасное для жизни состояние,
которое может развиваться у пациентов с болезнью Стилла. При возникновении или
подозрении на MAS необходимо как можно раньше начать обследование и
соответствующее лечение. Врачам следует внимательно относиться к симптомам
инфекции или обострению болезни Стилла, поскольку они являются триггерами MAS.
Доступные данные о возможности продолжать лечение препаратом Кинерет®
при тяжелых инфекциях у пациентов с болезнью Стилла ограничены. В случае
продолжения терапии препаратом Кинерет® на фоне тяжелых инфекций для
снижения риска обострения болезни, необходимо проводить строгий контроль
состояния пациента.
Пациенты
с болезнью Стилла имеют повышенный риск развития MAS. В ходе
пострегистрационного применения препарата были получены сообщения о случаях
развития MAS у пациентов с болезнью Стилла, получавших лечение препаратом
Кинерет®. В ходе КИ болезни Стилла, спонсированных компанией, случаи
MAS не были зарегистрированы. В долгосрочных неинтервенционных исследованиях
безопасности у 306 пациентов детского возраста с болезнью Стилла не сообщалось
о повышении частоты MAS во время и непосредственно после лечения препаратом
Кинерет®. Частота проявления MAS составляет 2,4 случая на 100
пациенто-лет, что соответствует ожидаемым значениям для пациентов детского
возраста с болезнью Стилла. Причинно-следственная связь между терапией
препаратом Кинерет® и развитием MAS не установлена.
Врачи
должны соблюдать осторожность при введении препарата Кинерет®
пациентам, имеющим в анамнезе рецидивирующие инфекции или предрасполагающие
факторы, которые могут привести к развитию инфекции.
Безопасность
применения препарата Кинерет® у пациентов с латентным туберкулезом
неизвестна. Имеются сообщения о туберкулезе у пациентов, получивших несколько
курсов лечения биологическими противовоспалительными препаратами. Перед
назначением препарата Кинерет® пациентов необходимо проверять на
наличие латентного туберкулеза. Имеющиеся медицинские рекомендации также должны
быть приняты во внимание.
Другие
виды противоревматической терапии ассоциировались с реактивацией гепатита В.
Скрининг на вирусный гепатит должен быть выполнен в соответствии с
опубликованными руководствами перед началом терапии препаратом Кинерет®.
Почечная недостаточность
Препарат
Кинерет® выводится путем клубочковой фильтрации и последующего
метаболизма в почечных канальцах. Соответственно, плазменный клиренс препарата
Кинерет® снижается при ухудшении функции почек.
Для
пациентов с почечной недостаточностью легкой степени тяжести (КК
60–89 мл/мин) коррекция дозы не требуется. У пациентов с почечной
недостаточностью средней степени тяжести (КК 30–59 мл/мин) препарат
Кинерет® следует применять с осторожностью. У пациентов с тяжелой
почечной недостаточностью (КК <30 мл/мин) или терминальной почечной
недостаточностью, включая пациентов на диализе, следует предусмотреть введение
назначенной дозы препарата Кинерет® через день.
Нейтропения
Применение
препарата Кинерет® было, как правило, связано с нейтропенией (ANC
<1,5 × 109/л) в плацебо-контролируемых КИ при
другом показании, но случаи нейтропении также были отмечены у пациентов с CAPS
и болезнью Стилла. Терапию препаратом Кинерет® не следует начинать у
пациентов с нейтропенией (ANC <1,5 × 109/л). Рекомендуется
проводить оценку количества нейтрофилов до начала лечения и во время применения
препарата Кинерет® ежемесячно в течение первых 6 месяцев лечения и
далее — каждые 3 месяца. Пациентам, у которых развивается нейтропения (ANC
<1,5 × 109/л), следует проводить тщательный контроль
количества нейтрофилов, и лечение препаратом Кинерет® у них должно
быть прекращено. Оценку безопасности и эффективности применения препарата
Кинерет® у пациентов с нейтропенией не проводили.
Нежелательные явления со стороны легких
Во
время пострегистрационного применения препарата отмечены случаи
интерстициального заболевания легких, легочного альвеолярного протеиноза и
легочной гипертензии, в основном, у детей с болезнью Стилла, получавших лечение
ингибиторами ИЛ-6 и ИЛ-1, включая препарат Кинерет®. Пациенты с
трисомией по 21-й хромосоме, как оказалось, представлены в большем количестве.
В ходе КИ болезни Стилла, спонсированных компанией, не было зарегистрировано
подобных случаев. В долгосрочных неинтервенционных исследованиях безопасности у
306 пациентов детского возраста с болезнью Стилла у одного пациента наблюдалось
серьезное легочное явление — неуточненное интерстициальное заболевание легких.
Пациентов с легочным альвеолярным протеинозом и легочной гипертензией в
исследовании не зарегистрировали. Причинно-следственная связь между применением
препарата Кинерет® и НЯ со стороны легких не установлена.
Лекарственно-индуцированная реакция с эозинофилией и
системными симптомами (DRESS)
Лекарственно-индуцированная
реакция с эозинофилией и системными симптомами (DRESS) наблюдалась у пациентов,
получавших лечение препаратом Кинерет®, в редких случаях, главным
образом — у пациентов с системным ювенильным идиопатическим артритом (SJIA).
Пациентам с DRESS может потребоваться госпитализация ввиду возможности
летального исхода. При появлении признаков и симптомов DRESS и отсутствии
обоснования иной причины, следует прекратить прием препарата Кинерет®
и назначить соответствующее лечение.
Иммуносупрессия
Влияние
лечения препаратом Кинерет® на уже имеющуюся злокачественную опухоль
не изучали. Поэтому, не рекомендуется назначать препарат Кинерет8 пациентам с
уже имеющимися злокачественными опухолями.
Вакцинация
Данные
плацебо-контролируемого КИ (n
= 126) не показали различий в выработке антител к столбняку в ответ на введение
дифтерийно-столбнячной вакцины пациентам, получавшим препарат Кинерет®
и плацебо. Данные о результатах вакцинации при введении других инактивированных
антигенов пациентам на фоне лечения препаратом Кинерет® отсутствуют.
Нет
данных и о реакциях на применение живых вакцин или о вторичной передаче
инфекции с живыми вакцинами пациентам, получающим Кинерет®. Таким
образом, живые вакцины не следует назначать пациентам на фоне лечения
препаратом Кинерет®.
Применение у пожилых пациентов (≥65 лет)
В
рамках КИ по показаниям, отличным от CAPS, FMF и болезни Стилла, препарат
Кинерет® изучали у 752 пациентов в возрасте >65 лет, в том числе,
у 163 пациентов в возрасте ≥75 лет. В целом, не было обнаружено
различий в безопасности и эффективности препарата у пациентов пожилого и более
молодого возраста. Опыт лечения пожилых пациентов с CAPS, FMF и болезнью Стилла
ограничен. Поскольку у пожилых заболеваемость инфекциями более высокая, во
время лечения этих пациентов следует соблюдать осторожность.
Вспомогательные вещества
Данный
препарат содержит менее 1 ммоль натрия (23 мг) на каждые 100 мг
воды, что практически означает отсутствие натрия в препарате.
Влияние на способность управлять автотранспортными
средствами, механизмами
Препарат
Кинерет® не оказывает влияния на способность управлять транспортными
средствами и работу с механизмами, требующими повышенной концентрации внимания.
Форма выпуска
Раствор для подкожного введения, 150 мг/мл
0,67 мл
раствора (100 мг) в градуированном предварительно заполненном шприце,
состоящем из стеклянного цилиндра (боросиликатное стекло I типа USP) со
встроенной нержавеющей стальной иглой для подкожного введения (размер 29G),
закрытой эластомерным колпачком с дополнительным внешним жестким
полипропиленовым колпачком, поршня и плунжера.
7
градуированных предварительно заполненных шприцев помещают в ПВХ блистер
(поддон). 1 блистер (поддон) вместе с инструкцией по применению помещают в
картонную пачку (вторичную упаковку) — индивидуальная упаковка.
4
картонные пачки, содержащие по 7 шприцев, упакованы в картонную коробку
(третичную упаковку) — групповая упаковка.
На
каждую картонную пачку и картонную коробку наклеивают прозрачную защитную
этикетку для контроля первого вскрытия.
Условия отпуска из аптек
Условия хранения
Хранить
при температуре от 2 до 8 °С в оригинальной упаковке для защиты от
воздействия света.
Не
замораживать.
Храните
в местах, недоступных для детей.
Препарат
Кинерет® допустимо хранить в течение 12 часов при температуре
не выше 25 °С.
По
истечению этого периода препарат необходимо выбросить.
Срок годности
3
года.
Не
использовать после истечения срока годности.
Состав
Главный функциональный компонент – анакинра.
Прочие составляющие: лимонная кислота, безводный хлорид натрия, дигидрат эдетата натрия, полисорбат 80, гидроксид натрия, вода для инъекций.
Форма выпуска
Медикамент имеет форму раствора для инъекций. Поставляется в упаковках по 7 наполненных шприцов в каждой.
Фармакологическое действие
Медпрепарат конкурентно ингибирует связывание с рецептором интерлейкина I типа, тем самым устраняя биологическую активность интерлейкина-1 и интерлейкина-1.
Фармакокинетика
Биодоступность после парентерального введения составляет 95%. Максимальная концентрация достигается через 3-7 часов.
Выводится через почки.
Показания к применению
Кинерет показан при нескольких состояниях, таких как: ревматоидный артрит в сочетании с метотрексатом, синдромы периодической лихорадки у детей с массой тела не менее 10 кг, подростки и взрослые, криопирин-зависимые периодические синдромы, семейная средиземноморская лихорадка в сочетании с колхицином, болезнь Стилла, детям с массой тела не менее 10 кг, подросткам и взрослым.
Противопоказания
Ограничениями к применению медикамента являются аллергия на составляющие вещества и нейтропения.
Побочные действия
Наиболее частые побочные эффекты Kineret (наблюдаемые более чем у 1 пациента из 10) — это головная боль и реакции в месте инъекции (покраснение, синяки, боль и воспаление), а также повышение уровня холестерина в крови.
Совместимость с другими медикаментами
Хотя не было взаимодействия между препаратом и другими лекарствами, включая НПВП, глюкокортикостероиды или противоревматические препараты, модифицирующие заболевание (DMARD), не стоит использовать Кинерет в сочетании с антагонистами фактора некроза опухоли (TNF) (другими лекарствами, применяемыми при ревматоидном артрите).
Применение и дозы
Суточная доза зависит от состояния здоровья и веса пациента. У взрослых она составляет 100 мг, у детей 1-2 мг на килограмм массы тела в сутки.
Передозировка
На сегодняшний день о случаях передозировки не сообщалось.
Особые указания
Медикамент увеличивает риск различных инфекций, в том числе при тяжелом течении болезни. Лечение не следует начинать у больных с активными инфекциями, а лечение у людей, у которых развилась инфекция, необходимо прекращать.
Применение при беременности и кормлении грудью
Назначение медикамента запрещено.
Влияние на способность к вождению автотранспорта и управлению механизмами
Особые меры предосторожности не требуются.
Условия продажи
По назначению доктора.
Условия хранения
В сухом, прохладном месте, с ограниченным доступом для детей.
Ингибитор фактора некроза опухоли альфа (ФНОα). Фактор некроза опухоли (ФНО) является основным цитокином, поддерживающим воспалительный процесс при ревматоидном артрите. Повышение концентрации ФНО также обнаружено в синовиальных оболочках и псориатических бляшках у пациентов с псориатическим артритом, а также в плазме и синовиальных тканях пациентов с анкилозирующим спондилитом. Инфильтрация воспалительными клетками, включая Т-клетки пораженных участков кожи при бляшечном псориазе, приводит к повышению в них концентрации ФНО по сравнению с кожей, не вовлеченной в патологический процесс. Этанерцепт является конкурентным ингибитором связывания ФНО с его рецепторами на поверхности клетки, и, таким образом, ингибирует биологическую активность ФНО. ФНО и лимфотоксин относятся к провоспалительным цитокинам, которые связываются с двумя четко различимыми рецепторами фактора некроза опухоли (ФНОР) на поверхности клетки: 55-килодальтон (р55) и 75-килодальтон (р75). Оба ФНОР присутствуют в организме в связанной с мембраной и свободной форме. Растворимые ФНОР регулируют биологическую активность ФНО.
ФНО и лимфотоксин существуют преимущественно как гомотримеры, их биологическая активность зависит от перекрестной сшивки ФНОР, находящихся на поверхности клетки. Димерные растворимые рецепторы, такие как этанерцепт, имеют большее сродство к ФНО, чем мономерные рецепторы, и, поэтому являются значительно более сильными конкурентными ингибиторами связывания ФНО с их клеточными рецепторами. Кроме того, использование области Fc-фрагмента иммуноглобулина, как элемента связывания в структуре димерного рецептора, удлиняет Т1/2 из сыворотки.
Механизм действия
Значительная часть патологических изменений в суставах при ревматоидном артрите и анкилозирующем спондилите, а также изменения кожи в виде псориатических бляшек возникают за счет воздействия провоспалительных молекул, вовлеченных в систему, контролируемую ФНО. Механизм действия этанерцепта, по-видимому, заключается в конкурентном ингибировании связывания ФНО с рецепторами ФНО на поверхности клетки. Таким образом, этанерцепт предупреждает клеточный ответ, опосредованный ФНО, способствуя биологической инактивации ФНО. Этанерцепт также может модулировать биологические ответы, контролируемые дополнительными молекулами, передающими сигнал по нисходящей (например, цитокины, молекулы адгезии или протеиназы). И эти ответы могут либо стимулировать, либо контролировать ФНО.
Клиническая эффективность и безопасность
В данном разделе представлены данные, полученные в ходе 4 рандомизированных контролируемых исследований у взрослых пациентов с ревматоидным артритом, 1 исследования у взрослых пациентов с псориатическим артритом, 1 исследования у взрослых пациентов с анкилозирующим спондилитом, 1 исследования у взрослых пациентов с нерентгенографическим аксиальным спондилоартритом, 4 исследований у взрослых пациентов с бляшечным псориазом, 3 исследований у пациентов с ювенильным идиопатическим артритом и 1 исследования у детей с бляшечным псориазом.
Взрослые пациенты с ревматоидным артритом
Оценку эффективности препарата Энбрел проводили в рандомизированном двойном слепом плацебо-контролируемом исследовании. В данном исследовании принимали участие 234 взрослых пациента с ревматоидным артритом в активной фазе, у которых терапия как минимум одним, но не более чем четырьмя болезнь-модифицирующими противоревматическими препаратами (DMARD) была неэффективной. В данном исследовании в течение 6 последовательных месяцев 2 раза в неделю вводили п/к либо Энбрел в дозе 10 мг или 25 мг, либо плацебо. Результаты данного контролируемого исследования выражались в виде процента улучшения течения ревматоидного артрита с использованием для этой цели критериев эффективности Американской коллегии ревматологов (АCR).
Показатели ответа на лечение по критериям ACR20 и ACR50 через 3 и 6 мес были выше у пациентов, получавших Энбрел, чем у пациентов, получавших плацебо (ACR20: Энбрел — 62% и 59%, плацебо — 23% и 11% через 3 и 6 мес соответственно; ACR50: Энбрел — 41% и 40%, плацебо — 8% и 5% через 3 и 6 мес, соответственно; р<0.01 Энбрел vs плацебо во всех временных точках как для ACR20, так и для ACR50). Приблизительно 15% пациентов, получавших Энбрел, достигли ответа ACR70 через 3 и 6 мес по сравнению с менее чем 5% пациентов в группе плацебо. У пациентов, получавших Энбрел, клинический ответ, как правило, проявлялся через 1-2 недели после начала лечения и практически всегда отмечался к 3 мес. Была выявлена зависимость эффекта от дозы препарата; эффект дозы 10 мг носил промежуточный (между плацебо и дозой 25 мг) характер. Энбрел значительно превосходил плацебо по всем компонентам критериев ACR, а также по другим показателям активности ревматоидного артрита, не включенным в критерии ответа ACR (например, утренняя скованность). В ходе исследования каждые 3 мес производилось заполнение опросника оценки состояния здоровья (HAQ), который включал в себя подразделы оценки нарушения жизнедеятельности, жизненного тонуса, умственного здоровья, общего состояния здоровья и состояния здоровья, связанного с артритом. У пациентов, получавших Энбрел, было отмечено улучшение по всем подразделам опросника оценки состояния здоровья (HAQ) в 3 и 6 мес по сравнению с пациентами контрольной группы.
После прекращения терапии препаратом Энбрел в течение месяца возможно обострение заболевания. По данным открытых исследований эффективность повторного назначения препарата в течение 24 мес после прекращения терапии сравнима с эффективностью препарата у больных, не прерывавших лечение. В открытых расширенных исследованиях с участием пациентов, непрерывно получавших Энбрел, продолжительный стойкий ответ наблюдался в течение 10 лет.
Сравнение эффективности препарата Энбрел и метотрексата проводили в рандомизированном исследовании с активным контролем с рентгенографической оценкой в слепом режиме в первичной конечной точке. В исследовании принимало участие 632 пациента с активным ревматоидным артритом (с длительностью заболевания <3 лет), ранее не получавших метотрексат. Энбрел в дозах 10 мг или 25 мг вводился п/к 2 раза в неделю в течение 24 мес. В течение первых 8 недель исследования доза метотрексата повышалась с 7.5 мг в неделю до максимальной — 20 мг в неделю, применение которой продолжалось до 24 мес. Клиническое улучшение, включая начало действия препарата в течение 2 недель применения Энбрела в дозе 25 мг, было схоже с клиническим улучшением, наблюдаемым в предыдущих исследованиях, и сохранялось до 24 мес. В начале исследования у пациентов имелась умеренная степень нарушения жизнедеятельности, средние значения баллов опросника HAQ варьировали от 1.4 до 1.5. Терапия препаратом Энбрел в дозе 25 мг приводила к существенному улучшению через 12 мес, причем приблизительно у 44% пациентов сумма баллов по опроснику HAQ достигла нормального значения (менее 0.5). Данный клинический результат сохранялся на 2 году исследования.
В данном исследовании оценка структурных повреждений суставов выполнялась рентгенологически и выражалась в виде изменения значения общего счета Шарпа (TSS) и его компонентов, количества эрозии и сужений суставной щели (JSN). Рентгенографическое исследование кистей рук/запястий и стоп проводилось в исходном состоянии, а также через 6, 12 и 24 мес. Энбрел в дозе 10 мг систематически оказывал меньшее влияние на структурные повреждения по сравнению с дозой 25 мг. Эффективность препарата Энбрел в дозе 25 мг значительно превышала таковую метотрексата по динамике количества эрозий в 12 и 24 мес. Различия между метотрексатом и препаратом Энбрел в дозе 25 мг в TSS и JSN не были статистически значимыми.
В другом двойном слепом рандомизированном исследовании с активным контролем проводили сравнение клинической эффективности, безопасности и рентгенологического прогрессирования ревматоидного артрита при монотерапии препаратом Энбрел (в дозе 25 мг 2 раза в неделю), монотерапии метотрексатом (в дозе от 7.5 до 20 мг в неделю, медиана дозы препарата составляла 20 мг) и совместном применении препарата Энбрел + метотрексат у 682 взрослых пациентов с активным ревматоидным артритом с продолжительностью заболевания от 6 мес до 20 лет (в среднем 5 лет), у которых ответ как минимум на 1 болезнь-модифицирующий противоревматический препарат (DMARD), кроме метотрексата, был недостаточен.
У пациентов, принимавших Энбрел в комбинации с метотрексатом, наблюдался более выраженный ответ по ACR20, 50 и 70 и улучшение индексов по шкале DAS и HAQ на 24 и 52 неделе, по сравнению с пациентами обеих групп, получавших монотерапию (полученные результаты представлены в таблице ниже). Значительное превосходство применения комбинации Энбрела и метотрексата перед монотерапией данными лекарственными средствами также отмечалось и через 24 мес.
Результаты оценки клинической эффективности через 12 мес: сравнение Энбрел vs Метотрексат vs комбинация Энбрел+метотрексат у пациентов с ревматоидным артритом с длительностью заболевания от 6 мес до 20 лет
| Конечная точка | Метотрексат (n=228) | Энбрел (n=223) | Энбрел+Метотрексат (n=231) |
| Ответ по ACRа | |||
| ACR20 | 58.8% | 65.5% | 74.5%t,φ |
| ACR50 | 36.4% | 43% | 63.2%t,φ |
| ACR70 | 16.7% | 22% | 39.8%t,φ |
| Индекс активности заболевания (DAS) | |||
| Индекс в исходном состоянииб | 5.5 | 5.7 | 5.5 |
| Индекс на 52-й неделеб | 3 | 3 | 2.3t,φ |
| Ремиссияв | 14% | 18% | 37%t,φ |
| HAQ | |||
| Исходное состояние | 1.7 | 1.7 | 1.8 |
| 52-я неделя | 1.1 | 1 | 0.8t,φ |
а пациентов, не завершивших 12-месячный период участия в исследовании, рассматривали как пациентов, не ответивших на лечение.
б значения индекса активности заболевания (DAS) представлены в виде средних величин.
в ремиссия определялась как DAS <1.6.
Парное сравнение значений р: t=р<0.05 для сравнения Энбрел+метотрексат vs. метотрексат и φ=р<0.05 для сравнения Энбрел+метотрексат vs Энбрел.
Рентгенологическое прогрессирование через 12 мес было существенно менее выраженными в группе Энбрела, по сравнению с группой метотрексата, в то время как комбинированная терапия вызывала более выраженное замедление темпов рентгенологического прогрессирования.
Значительное превосходство применения комбинации препарата Энбрел и метотрексата перед монотерапией данными лекарственными средствами также отмечалось и через 24 мес. Спустя 24 мес также наблюдалось значительное превосходство монотерапии препаратом Энбрел над монотерапией метотрексатом.
В анализе, в котором всех пациентов, прекративших участие в исследовании по любой причине, рассматривали как пациентов с прогрессированием заболевания, процентная доля пациентов без прогрессирования (изменение значения общего счета Шарпа [TSS] ≤0.5) через 24 мес превышала таковую в группе, принимавшей Энбрел в комбинации с метотрексатом по сравнению с группами, получавшими монотерапию Энбрелом и метотрексатом (62%, 50% и 36%, соответственно; р<0.05). Отличие между монотерапией Энбрелом и метотрексатом также было статистически значимым (р<0.05). Среди пациентов, которые полностью прошли 24-месячный курс терапии в исследовании, частота отсутствия прогрессирования заболевания составляла 78%, 70% и 61% соответственно.
Оценку безопасности применения препарата Энбрел в дозе 50 мг (2 п/к инъекции по 25 мг) 1 раз в неделю проводили в двойном слепом плацебо-контролируемом исследовании с участием 420 пациентов с активным ревматоидным артритом. В данном исследовании 53 пациента получали плацебо, 214 пациентов получали Энбрел в дозе 50 мг 1 раз в неделю и 153 пациента — Энбрел в дозе 25 мг 2 раза в неделю. Профили безопасности и эффективности двух режимов применения препарата Энбрел на 8 неделе были сопоставимыми по их влиянию на симптомы ревматоидного артрита; данные на 16 неделе продемонстрировали отсутствие сопоставимости (не меньшей эффективности) между двумя режимами терапии.
Взрослые пациенты с псориатическим артритом
Оценка эффективности препарата Энбрел проводилась в рандомизированном двойном слепом плацебо-контролируемом исследовании с участием 205 пациентов в возрасте от 18 до 70 лет с псориатическим артритом в активной фазе (>3 опухших суставов и >3 болезненных при пальпации суставов) как минимум в одной из следующих форм: (1) артрит дистальных межфаланговых суставов (АДМФС), (2) полиартикулярный артрит (отсутствие ревматоидных узелков и наличие псориаза), (3) мутилирующий артрит, (4) асимметричный псориатический артрит или (5) псориатический спондилит. У пациентов также имелся бляшковидный псориаз с очагом поражения, имеющим размер >2 см в диаметре. Пациенты ранее получали НПВС (86%), DMARD (80%) и кортикостероиды (24%). Пациенты, принимавшие в начале исследования метотрексат (в стабильной дозе в течение >2 мес) могли продолжать применение метотрексата в стабильной дозе <25 мг/неделю. Препарат Энбрел в дозе 25 мг (на основании результатов исследований по определению диапазона доз у пациентов с ревматоидным артритом) или плацебо вводили п/к 2 раза в неделю в течение 6 мес. По окончании двойного слепого исследования пациенты могли входить в долгосрочное открытое исследование дополнительного лечения продолжительностью до 2 лет.
Клинический ответ выражался в виде процента пациентов, достигших ACR20, 50 и 70 и процента улучшения по критериям ответа на терапию псориатического артрита (PsARC). Полученные результаты представлены в таблице ниже.
Терапевтический ответ у пациентов с псориатическим артритом в плацебо-контролируемом исследовании
| Терапевтический ответ при псориатическом артрите | Процент пациентов | |
| Плацебо (n=104) | Энбрела (n=101) | |
| ACR20 | ||
| 3 месяц | 15 | 59б |
| 6 месяц | 13 | 50б |
| ACR50 | ||
| 3 месяц | 4 | 38б |
| 6 месяц | 4 | 37б |
| ACR70 | ||
| 3 месяц | 0 | 11б |
| 6 месяц | 1 | 9в |
| PsARC | ||
| 3 месяц | 31 | 72б |
| 6 месяц | 23 | 70б |
а Энбрел в дозе 25 мг п/к 2 раза в неделю.
б р <0.001, Энбрел vs плацебо.
в р <0.01, Энбрел vs плацебо.
У пациентов с псориатическим артритом, получавших Энбрел, клинический ответ наблюдался во время первого визита (4 неделя) и сохранялся в течение 6 мес терапии. Энбрел значительно превосходил плацебо по всем показателям активности заболевания (р<0.001), причем данный ответ был одинаковым при проведении сопутствующей терапии метотрексатом, так и без нее. Качество жизни пациентов с псориатическим артритом оценивали в каждой временной точке, используя индекс нарушения жизнедеятельности oпpocника HAQ. У пациентов с псориатическим артритом, получавших Энбрел, в каждой временной точке отмечалось значительное улучшение индекса нарушения жизнедеятельности по сравнению с плацебо (р<0.001).
В исследовании псориатического артрита проводилась оценка рентгенологических изменений. Рентгенография кистей и запястий проводилась в исходном состоянии, а также через 6, 12 и 24 мес. Показатели модифицированного общего счета Шарпа (TSS) через 12 мес представлены в таблице ниже. В анализе, в котором всех пациентов, прекративших участие в исследовании по любой причине, рассматривали как пациентов с прогрессированием заболевания, процентная доля пациентов без прогрессирования (изменение общего счета Шарпа [TSS] ≤0.5) через 12 мес была больше в группе, принимавшей Энбрел, по сравнению с группой плацебо (73% vs 47%, соответственно; р≤0.001). Влияние препарата Энбрел на рентгенологическое прогрессирование заболевания сохранялось у пациентов, продолжавших лечение в течение второго года. У пациентов с полиартикулярным симметричным артритом наблюдалось замедление прогрессирования поражения периферических суставов.
Среднее (СО) годовое изменение общего счета Шарпа относительно исходной величины
| Время | Плацебо (n=104) | Этанерцепт (n=101) |
| 12 мес | 1 (0.29) | -0.03 (0.09)а |
СО — стандартная ошибка.
а р=0.0001.
В ходе двойного слепого периода терапия препаратом Энбрел приводила к улучшению функционального статуса и данный эффект сохранялся при длительном (до 2 лет) применении препарата.
Данных об эффективности препарата Энбрел у пациентов с псориатическим спондлитом и мутилирующей формой псориатической артропатии недостаточно, что обусловлено небольшим количеством пациентов, участвовавших в исследовании.
У пациентов с псориатическим артритом исследование с использованием схемы приема препарата в дозе 50 мг 1 раз в неделю не проводилось. Данные об эффективности схемы применения препарата 1 раз в неделю в этой популяции пациентов были получены в исследовании у пациентов с анкилозирующим спондилитом.
Взрослые пациенты с анкилозирующим спондилитом
Оценку эффективности препарата Энбрел при анкилозирующем спондилите проводили в 3 рандомизированных двойных слепых исследованиях, сравнивавших применение препарата Энбрел в дозе 25 мг 2 раза в неделю с плацебо. Всего 401 пациент был включен в данные исследования, из которых 203 пациента принимали Энбрел. Самое крупное из этих исследований (n=277) включало пациентов в возрасте от 18 до 70 лет с активным анкилозирующим спондилитом, критериями которого являлись количество баллов ≥30 по визуальной аналоговой шкале (ВАШ) для средней продолжительности и интенсивности утренней скованности плюс ≥30 баллов по ВАШ, по крайней мере, для 2 из 3 следующих параметров: общая оценка состояния пациента; среднее количество баллов по ВАШ для ночной и общей боли в спине; средний балл для 10 вопросов по функциональному индексу анкилозирующего спондилита (BASFI). Пациенты, получавшие DMARD, НПВС или кортикостероиды, могли продолжать их применение в стабильных дозах. Пациенты с полным анкилозом позвоночника не включались в данное исследование. 138 пациентам в течение 6 мес п/к 2 раза в неделю вводили препарат Энбрел в дозе 25 мг (на основании результатов исследований по определению диапазона доз у пациентов с ревматоидным артритом) или плацебо.
Основным показателем эффективности (ASAS20) было ≥20% улучшение, по крайней мере, по 3 из 4 разделов шкалы оценки анкилозирующего спондилита (ASAS) (общая оценка заболевания пациентом, боль в спине, индекс BASFI и воспаление) и отсутствие ухудшения по остальным доменам шкалы. Ответы по шкалам ASAS50 и 70 определяли по тем же критериям — 50% или 70% улучшение, соответственно.
Лечение препаратом Энбрел по сравнению с плацебо приводило к значительным улучшениям по показателям ASAS20, ASAS50 и ASAS70 уже через 2 недели после начала терапии.
Терапевтический ответ у пациентов с анкилозирующим спондилитом в плацебо-контролируемом исследовании
| Терапевтический ответ при анкилозирующем спондилите | Процент пациентов | |
| Плацебо (n=139) | Энбрел (n=138) | |
| ASAS20 | ||
| 2 недели | 22 | 46а |
| 3 мес | 27 | 60а |
| 6 мес | 23 | 58а |
| ASAS50 | ||
| 2 недели | 7 | 24а |
| 3 мес | 13 | 45а |
| 6 мес | 10 | 42а |
| ASAS70 | ||
| 2 недели | 2 | 12б |
| 3 мес | 7 | 29б |
| 6 мес | 5 | 28б |
а р<0.001, Энбрел vs плацебо
б р=0.002, Энбрел vs плацебо
У пациентов с анкилозирующим спондилитом, получавших Энбрел, клинический ответ наблюдался во время первого визита (2 недели) и сохранялся в течение 6 мес терапии, причем выраженность этого ответа у пациентов при сопутствующем приеме лекарственных средств в начале исследования и без него была одинаковой.
Аналогичные результаты были получены в 2 меньших исследованиях пациентов с анкилозирующим спондилитом.
В четвертом двойном слепом плацебо-контролируемом исследовании с участием 356 пациентов с активным анкилозирующим спондилитом проводили оценку безопасности и эффективности применения препарата Энбрел в дозе 50 мг (2 п/к инъекции по 25 мг) 1 раз в неделю в сравнении с применением препарата в дозе 25 мг 2 раза в неделю. Профили безопасности и эффективности препарата при его применении в дозе 50 мг 1 раз в неделю и 25 мг 2 раза в неделю были одинаковыми.
Пациенты с нерентгенографическим аксиальным спондилоартритом
Оценку эффективности препарата Энбрел у пациентов с нерентгенографическим аксиальным спондилоартритом (Нр-АСпа) выполняли в рандомизированном двойном слепом 12-недельном плацебо-контролируемом исследовании. В исследование было включено 215 пациентов (модифицированная популяция пациентов, включенных в исследование) с активным Нр-АСпа (в возрасте от 18 до 49 лет). При включении пациенты должны были соответствовать критериям американской классификации аксиального спондилоартрита, но не соответствовали критериям модифицированной шкалы Нью-Йорк для анкилозирующего спондилоартрита (АС). Также одним из критериев включения было наличие неадекватного ответа или непереносимости двух и более НПВС. В течение двойного слепого периода пациентам назначали Энбрел в дозе 50 мг 1 раз в неделю или плацебо в течение 12 недель. Первичным критерием эффективности (по шкале оценки при анкилозирующем спондилите ASAS40) было улучшение на 40% по не менее 3 из 4 категорий шкалы, а также отсутствие ухудшения по 4 блоку. Воспаление оценивали по результатам МРТ крестцово-подвздошного отдела и позвоночника в начале исследования и через 12 недель. После двойного слепого периода наступал открытый период, во время которого все пациенты получали Энбрел в дозе 50 мг 1 раз в неделю в течение дополнительных 92 недель.
По сравнению с плацебо лечение препаратом Энбрел привело к значительному улучшению ASAS40, ASAS20 и ASAS5/6, также отмечались значительные улучшения по ASAS частичная ремиссия и индекса активности заболевания анкилозирующим спондилитом (BASDAI50). В таблице ниже приведены результаты на 12 неделе исследования.
Результат для оценки эффективности в плацебо-контролируемом исследовании Нр-АСпа: % пациентов, достигших конечной точки
| Двойные слепые клинические ответы через 12 недель | Плацебо (n=106-109*) | Энбрел (n=103-105*) |
| ASAS**40 | 15/7 | 32.4b |
| ASAS20 | 36.1 | 52.4c |
| ASAS5/6 | 10.4 | 33а |
| ASAS частичное восстановление | 11.9 | 24.8c |
| BASDAI***50 | 23.9 | 43.8b |
* Для отдельных пациентов не получено данных для всех конечных точек
** ASAS = шкала оценки при анкилозирующем спондилите
*** Индекс активности заболевания анкилозирующим спондилитом
а: р<0.001, b: р<0.01 и c: р<0.05 между Энбрелом и плацебо соответственно
Через 12 недель отмечалось значительное улучшение балла по шкале SPARCC (консорциум Канады по изучению спондилоартрита) для крестцово-подвздошного отдела, оценку результатов проводили по МРТ пациентов, получавших Энбрел. Скорректированное среднее изменение от исходного значения составило 3.8 для группы Энбрел (n=95) по сравнению с 0.8 в группе плацебо (n=105) (р<0.001).
Установлено, что применение препарата Энбрел позволяет получить значительное улучшение по сравнению с исходным значением и плацебо через 12 недель по таким показателям, как качество жизни, связанное с состоянием здоровья, и оценка функционального статуса пациента, включая BASFI (функциональный индекс при анкилозирующем спондилите), балл для общего состояния организма EuroQol 5D и индекс физического здоровья SF-36.
У пациентов с Нр-АСпа, получающих терапию препаратом Энбрел, отмечали явные клинические ответы уже во время первого визита (2 недели), которые сохранялись в течение 24 недель лечения.
Взрослые пациенты с бляшковидным псориазом
Препарат Энбрел рекомендуется применять у пациентов согласно указаниям в разделе «Показания». Пациенты, не отвечавшие на лечение в целевой популяции, определялись при недостаточном ответе (индекс PASI <50 или общая оценка врача (РGА) хуже чем «удовлетворительная») или усугублении заболевания в ходе лечения, а также к ним относились пациенты, получавшие в надлежащей дозе каждый из трех основных видов системных препаратов в течение достаточно длительного периода времени, чтобы произвести оценку эффективности.
Оценка эффективности препарата Энбрел в сравнении с другими видами системной терапии у пациентов с умеренным или тяжелым псориазом (реагирующих на другие виды системной терапии) в исследованиях, непосредственно сравнивающих Энбрел с другими видами системной терапии, не проводилась. Вместо этого эффективность и безопасность препарата Энбрел оценивали в 4 рандомизированных двойных слепых плацебо-контролируемых исследованиях. Первичной конечной точкой оценки эффективности во всех 4 исследованиях была процентная доля пациентов в каждой группе лечения, достигавших PASI 75 (т.е., по меньшей мере, 75% улучшение индекса распространенности и тяжести псориаза относительно исходного уровня) на 2 неделе исследования.
Исследование 1 представляло собой исследование 2 фазы у пациентов в возрасте ≥18 лет с активным, но клинически стабильным бляшковидным псориазом с поражением ≥10% площади поверхности тела. 112 пациентов были рандомизированы на получение препарата Энбрел в дозе 25 мг (n=57) или плацебо (n=55) 2 раза в неделю в течение 24 недель.
В исследовании 2 проводили оценку 652 пациентов с хроническим бляшковидным псориазом, используя те же самые критерии включения, что и в исследовании 1 с добавлением минимального индекса распространенности и тяжести псориаза (PASI), равного 10 при скрининговом обследовании. Препарат Энбрел назначался в дозах 25 мг 1 раз в неделю, 25 мг 2 раза в неделю или 50 мг 2 раза в неделю в течение 6 последовательных месяцев. В течение первых 12 недель лечения пациенты в двойном слепом режиме получали плацебо или одну из трех вышеуказанных доз препарата Энбрел. Спустя 12 недель пациенты группы плацебо в слепом режиме начинали прием препарата Энбрел (в дозе 25 мг 2 раза в неделю); пациенты в активных группах лечения продолжали прием препарата до 24 недели в дозе, изначально полученной при рандомизации.
В исследовании 3 проводили оценку 583 пациентов и использовали критерии, аналогичные критериям включения в исследование 2. В этом исследовании пациенты получали Энбрел в дозе 25 мг или 50 мг либо плацебо 2 раза в неделю в течение 12 недель, после чего все пациенты в открытом режиме дополнительно получали Энбрел в дозе 25 мг 2 раза в неделю еще в течение 24 недель.
В исследовании 4 проводили оценку 142 пациентов и использовали критерии, аналогичные критериям включения в исследование 2 и 3. В данном исследовании пациенты получали Энбрел в дозе 50 мг или плацебо 1 раз в неделю в течение 12 недель, после чего все пациенты в открытом режиме еще в течение 12 недель получали Энбрел в дозе 50 мг 1 раз в неделю.
В исследовании 1 на 12 неделе процент пациентов, достигших индекса PASI 75, был статистически достоверно большим в группе, получавшей Энбрел (30%), чем в группе плацебо (2%) (р<0.0001). На 24 неделе 56% пациентов в группе, получавшей Энбрел, достигли индекса PASI 75 по сравнению с 5% пациентов в группе плацебо. Основные результаты исследований 2, 3 и 4 представлены ниже.
Терапевтический ответ у пациентов с псориазом в исследованиях 2, 3 и 4
| Ответ (%) | Исследование 2 | ||||
| Плацебо (n=166) 12 недель | Энбрел | ||||
| 25 мг 2 раза/нед. | 50 мг 2 раза/нед. | ||||
| (n=162) 12 недель | (n=162) 24 недели | (n=164) 12 недель | (n=164) 24 недели | ||
| PASI 50 | 14 | 58* | 70 | 74* | 77 |
| PASI 75 | 4 | 34* | 44 | 49* | 59 |
| DSGAб, чисто или практически чисто | 5 | 34* | 39 | 49* | 55 |
| Ответ (%) | Исследование 3 | Исследование 4 | ||||
| Плацебо (n=193) 12 недель | Энбрел | Плацебо (n=46) 12 недель | Энбрел | |||
| 25 мг 2 раза/нед., (n=196) 12 недель | 50 мг 2 раза/нед., (n=196) 12 недель | 50 мг 1 раз/нед., (n=96) 12 недель | 50 мг 1 раз/нед., (n=90) 24 неделиа | |||
| PASI 50 | 9* | 64* | 77* | 9 | 69* | 83 |
| PASI 75 | 3* | 34* | 49* | 2 | 38* | 71 |
| DSGAб,чисто или практически чисто | 39* | 39* | 57* | 4 | 39* | 64 |
* р<0.0001 по сравнению с плацебо.
а в исследованиях 2 и 4 на 24 неделе не проводили статистических сравнений с плацебо, поскольку пациенты группы,
первоначально получавшей плацебо, начинали получать Энбрел в дозе 25 мг 2 раза/нед. или 50 мг 1 раз/нед. с 13 по 24 неделю.
б шкала статистической глобальной оценки дерматологом. «Чисто» или «пратически чисто» — 0 или 1 по шкале от 0 до 5.
У пациентов с бляшковидным псориазом, получавших Энбрел, значимый клинический ответ наблюдался во время первого визита (2 недели) и сохранялся в течение 24 недель терапии по сравнению с плацебо.
В исследовании 2 также имелся период отмены исследуемого препарата, в течение которого пациенты, достигавшие как минимум на 24 неделе значения PASI 50 (улучшения на 50%), прекращали терапию. Пациенты, не получая лечение, находились под наблюдением врача с целью выявления синдрома отмены (PASI ≥150% исходного уровня) и для определения времени до рецидива заболевания (при утрате, по крайней мере, половины улучшения, достигнутого в период между исходным уровнем и 24 неделей). В течение периода отмены препарата симптомы псориаза постепенно возвращались, при этом медиана времени до рецидива заболевания составляла 3 мес. Случаев внезапного развития синдрома отмены и серьезных нежелательных явлений, связанных с псориазом, зарегистрировано не было. Были получены данные, свидетельствующие о пользе повторного лечения препаратом Энбрел пациентов, первоначально ответивших на лечение.
В исследовании 3 у большинства пациентов (77%), первоначально рандомизированных на получение препарата Энбрел в дозе 50 мг 2 раза в неделю, при снижении дозы препарата до 25 мг 2 раза в неделю на 12 неделе сохранялся ответ PASI 75 по 36 неделю включительно. У пациентов, получавших в ходе исследования препарат в дозе 25 мг 2 раза в неделю, ответ PASI 75 продолжал улучшаться с 12 по 36 недели.
В исследовании 4 на 12 неделе процент пациентов, достигших индекса PASI 75, был статистически достоверно большим в группе, получавшей Энбрел (38%), чем в группе плацебо (2%) (р<0.0001). У пациентов, получавших Энбрел в дозе 50 мг 1 раз в неделю в ходе всего исследования, клинический эффект продолжал расти, причем 71% пациентов достигли PASI 75 на 24 неделе.
В долгосрочных (продолжительностью до 34 мес) открытых исследованиях с непрерывным приемом препарата Энбрел, клинические ответы были устойчивы, а безопасность была сопоставимой с таковой в краткосрочных исследованиях.
Анализ данных клинического исследования не выявил каких-либо исходных характеристик заболевания, которые могли бы помочь клиницистам в выборе наиболее подходящего режима дозирования (интермитирующего или непрерывного). Следовательно, выбор интермиттирующей или непрерывной схемы терапии должен оставляться на усмотрение врача и основываться на индивидуальных потребностях пациента.
Антитела к этанерцепту
В сыворотке некоторых пациентов, получавших препарат, были обнаружены антитела к этанерцепту. Все эти антитела не являлись нейтрализующими антителами, и, как правило, их появление носило транзиторный характер. Корреляция между выработкой антител и клиническим ответом или нежелательными явлениями, по-видимому, отсутствует.
У пациентов, получавших этанерцепт в утвержденных дозах в клинических исследованиях до 12 мес, суммарная частота выработки антител против этанерцепта составляла приблизительно 6% у пациентов с ревматоидным артритом, 7.5% — у пациентов с псориатическим артритом, 2% — у пациентов с анкилозирующим спондилитом, 7% — у пациентов с псориазом, 9.7% — у пациентов с детским псориазом и 4.8% — у пациентов с ювенильным идиопатическим артритом.
Как и ожидалось, доля пациентов, у которых вырабатывались антитела к этанерцепту в долгосрочных исследованиях (продолжительностью до 3.5 лет) со временем увеличивается. Тем не менее, в связи с транзиторным характером данного явления, у пациентов с ревматоидным артритом и пациентов с псориазом частота выявления антител в каждой точке проведения оценки, как правило, была меньше 7%.
В долгосрочном исследовании псориаза, в котором пациенты получали препарат в дозе 50 мг 2 раза в неделю в течение 96 недель, частота выработки антител, наблюдаемая в каждой точке проведения оценки, составляла приблизительно до 9%.
Дети с ювенильным идиопатическим артритом
Оценку безопасности и эффективности препарата Энбрел проводили в исследовании, состоявшем из двух частей, с участием 69 детей с полиартикулярной формой ювенильного идиопатического артрита с различными типами манифестации заболевания (полиартрит, пауциартрит и системное начало). В исследование включались пациенты в возрасте от 4 до 17 лет с полиартикулярной формой ювенильного идиопатического артрита умеренной и высокой активности, которые трудно поддавались лечению, или у которых отмечалась непереносимость метотрексата. Пациенты продолжали получать стабильную дозу одного НПВС и/или преднизона (<0.2 мг/кг/сут или 10 мг максимум). В 1 части исследования все пациенты получали Энбрел в дозе 0.4 мг/кг (максимальная разовая доза 25 мг) п/к 2 раза в неделю. Во 2 части исследования пациенты с клиническим эффектом на 90 день были рандомизированы на продолжение приема препарата Энбрел или начало приема плацебо в течение 4 мес, после чего подвергались оценке на предмет выявления обострения заболевания. Для оценки эффективности терапии использовались критерии ACR Pedi 30 (≥30% улучшение, как минимум, по трем из шести и ≥30% ухудшение не более чем по одному из шести основных критериев ЮРА, включая количество пораженных суставов, ограничение в движении, общую оценку врачом и пациентом/родителями, оценку функциональных нарушений и СОЭ. Обострение заболевания определяли как ≥30% ухудшение по трем из шести основных критериев ЮРА и ≥30% улучшение не более чем по одному из шести основных критериев ЮРА, и минимум два пораженных сустава.
В 1 части исследования 51 пациент из 69 (74%) продемонстрировал клинический ответ и был включен во 2 часть. Во 2 части исследования у 6 из 25 (24%) пациентов, продолжавших получать Энбрел, было зарегистрировано обострение заболевания по сравнению с 20 из 26 (77%) пациентов, получавших плацебо (р=0.007). С начала 2 части исследования медиана времени до обострения заболевания составляла ≥116 дней для пациентов, получавших Энбрел, и 28 дней для пациентов, получавших плацебо. Из пациентов, продемонстрировавших клинический ответ через 90 дней и включенных во 2 часть исследования, некоторые и далее получали Энбрел и продолжали демонстрировать улучшение с 3 по 7 мес, в то время как пациенты, получавшие плацебо, не демонстрировали улучшения.
В открытом расширенном исследовании безопасности 58 детей из упомянутого выше исследования (в возрасте на момент включения от 4 лет) продолжили получать Энбрел до 10 лет. При долгосрочном применении частота серьезных нежелательных явлений и серьезных инфекций не увеличивалась.
Оценку долгосрочной безопасности монотерапии препаратом Энбрел (n=103), комбинации Энбрела и метотрексата (n=294) или монотерапии метотрексатом (n=197) проводили в течение 3 лет с участием 594 детей в возрасте от 2 до 18 лет с ювенильным идиопатическим артритом (из которых 39 детей были в возрасте от 2 до 3 лет). В целом, у пациентов, получавших этанерцепт, чаще регистрировались инфекции по сравнению с монотерапией метотрексатом (3.8 vs 2%), причем инфекции, связанные с применением этанерцепта, носили более тяжелый характер.
В другом открытом несравнительном исследовании 60 пациентов с распространенным олигоартритом (15 пациентов в возрасте от 2 до 4 лет, 23 пациента в возрасте от 5 до 11 лет и 22 пациента в возрасте от 12 до 17 лет), 38 пациентов с артритом, связанным с энтезитом (в возрасте от 12 до 17 лет) и 29 пациентов с псориатическим артритом (в возрасте от 12 до 17 лет) получали Энбрел в дозе 0.8 мг/кг (максимальная доза составляла 50 мг) 1 раз в неделю в течение 12 недель. Большинство пациентов каждого подтипа ЮИА отвечали критериям ACR Pedi 30 и демонстрировали клиническое улучшение по вторичным конечным точкам (таким как, количество болезненных при пальпации суставов и общая оценка врача). Профиль безопасности препарата был сопоставим с профилем безопасности в других исследованиях пациентов с ЮИА.
Исследования по оценке влияния продолжительной терапии препаратом Энбрел на пациентов с ювенильным идиопатическим артритом, у которых не наблюдалось терапевтического ответа в течение 3 мес после начала терапии препаратом Энбрел, не проводились. Кроме того, у пациентов с ЮИА не проводились исследования по оценке влияния прекращения применения препарата или снижения его рекомендуемой дозы после длительного применения.
Дети с бляшковидным псориазом
Оценку эффективности препарата Энбрел проводили в рандомизированном двойном слепом плацебо-контролируемом исследовании с участием 211 пациентов в возрасте от 4 до 17 лет с бляшечным псориазом средней и тяжелой степени (количество баллов по шкале РGА ≥3, включая BSA ≥10% и PASI ≥12). Пациенты, соответствовавшие критериям включения в данное исследование, ранее проходили фото- или системную терапию, или местная терапия была недостаточно эффективна.
Пациенты получали Энбрел в дозе 0.8 мг/кг (до 50 мг) или плацебо 1 раз в неделю в течение 12 недель. На 12 неделе исследования положительная динамика (например, PASI 75) отмечалась у большего количества пациентов, рандомизированных на получение препарата Энбрел, по сравнению с плацебо.
Исходы бляшковидного псориаза у детей на 12 неделе терапии
| Энбрел 0.8 мг/кг 1 раз в неделю (n=106) | Плацебо (n=105) | |
| PASI 75, n (%) | 60 (57%)а | 12 (11%) |
| PASI 50, n (%) | 79 (75%)а | 24 (23%) |
| sPGA «чисто» или «минимальные поражения», n (%) |
56 (53%)а | 14 (13%) |
sPGA — статическая глобальная оценка врача
а р<0.0001 по сравнению с плацебо.
По окончании 12-недельного двойного слепого периода лечения все пациенты дополнительно получали Энбрел в дозе 0.8 мг/кг (до 50 мг) 1 раз в неделю в течение 24 недель. Ответы, зарегистрированные в ходе открытого периода, были аналогичны ответам, наблюдавшимся в течение двойного слепого периода.
В течение периода рандомизированной отмены препарата у значительно большего количества пациентов, повторно рандомизированных на получение плацебо, регистрировался рецидив заболевания (утрата ответа PASI 75) по сравнению с пациентами, повторно рандомизированными в группу Энбрела. При продолжении терапии ответы сохранялись в течение 48 недель.
Оценку безопасности и эффективности долгосрочного применения препарата Энбрел в дозе 0.8 мг/кг (до 50 мг) 1 раз в неделю проводили в открытом расширенном клиническом исследования с участием 181 пациента в детском возрасте с бляшковидным псориазом в течение 2 лет, которое проводилось по окончании 48-недельного исследования, описанного выше. Опыт длительного применения препарата Энбрел, как правило, был сопоставим с таковым, полученным в первоначальном 48-недельном исследовании, и не выявил каких-либо новых сведений по безопасности.