Эврисди — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер
ЛП-006602
Торговое наименование
Эврисди®
Международное непатентованное или группировочное наименование
Рисдиплам
Лекарственная форма
Порошок для приготовления раствора для приема внутрь
Состав
Один флакон (2 г порошка для приготовления раствора для приема внутрь) содержит:
действующее вещество: рисдиплам – 60 мг;
вспомогательные вещества: маннитол – 1344.70 мг, изомальт – 237.25 мг, ароматизатор клубничный (мальтодекстрин кукурузный, крахмал кукурузный восковой модифицированный (Е1450), ароматизирующие компоненты) – 150.00 мг, винная кислота – 120.50 мг, натрия бензоат – 30.00 мг, макрогол/полиэтиленгликоль 6000 – 20.00 мг, сукралоза – 16.00 мг, аскорбиновая кислота – 14.10 мг, динатрия эдетата дигидрат – 7.45 мг.
1 мл приготовленного (восстановленного) раствора содержит 0.75 мг рисдиплама.
Описание
Порошок или порошок с комками от светло-желтого до желтого с сероватым или зеленоватым оттенком или светло-зеленого с желтоватым оттенком цвета (при выпуске). Порошок или порошок с комками, или скомковавшийся порошок от светло-желтого до желтого с сероватым или зеленоватым оттенком или светло-зеленого с желтоватым оттенком цвета (в течение срока годности).
Приготовленный (восстановленный) раствор
Прозрачный раствор от зеленовато-желтого до желтого цвета.
Фармакотерапевтическая группа
Прочие препараты для лечения заболеваний костно-мышечной системы.
Код АТХ
М09АХ10
Фармакологические свойства
Фармакодинамика
Механизм действия
Рисдиплам представляет собой модификатор сплайсинга предшественника матричной рибонуклеиновой кислоты (пре-мРНК) гена выживаемости двигательных нейронов 2 (SMN2), разработанный для лечения спинальной мышечной атрофии (СМА), причиной которой являются мутации в хромосоме 5q, что приводит к недостаточности белка SMN. Недостаточность функционального белка SMN является патофизиологическим механизмом развития СМА всех типов. Рисдиплам корректирует сплайсинг SMN2, сдвигая баланс с исключения экзона 7 на включение экзона 7 в транскрипте м-РНК. приводя к образованию функционального и стабильного белка SMN. Таким образом, рисдиплам лечит СМА путем увеличения и сохранения уровней функционального белка SMN.
Рисдиплам равномерно распределяется во всем организме, в том числе в центральной нервной системе (ЦНС), проникая через гематоэнцефалический барьер и соответственно приводя к увеличению уровня белка SMN в ЦНС и по всему организму. Концентрации рисдиплама в плазме и белка SMN в крови отражают распределение и фармакодинамические эффекты рисдиплама в тканях, а именно: мышечных тканях и тканях мозга.
Во всех клинических исследованиях применение рисдиплама приводило к устойчивому и длительному увеличению уровня белка SMN. В течение 4 недель после начала лечения медианный уровень белка SMN был более чем в 2 раза выше по сравнению с исходным значением, согласно измерениям в крови. Повышение уровня белка SMN сохранялось на протяжении периода лечения ≤2 лет у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА (см. подраздел «Клиническая эффективность»).
Клиническая эффективность
Эффективность препарата Эврисди® для лечения пациентов с манифестацией СМА в младенческом возрасте и пациентов с поздней манифестацией СМА оценивалось в 2 опорных клинических исследованиях FIREFISH и SUNFISH и подтверждено дополнительными данными, полученными в исследовании JEWELFISH. В целом результаты исследований подтверждают эффективность препарата Эврисди® у пациентов со СМА.
Манифестация СМА в младенческом возрасте
ВР39056 (FIREFISH) представляет собой открытое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов с симптоматической СМА 1 типа (у всех пациентов было генетически подтверждено заболевание с двумя копиями гена SMN2). Исследование состоит из двух частей. Часть 1 исследования FIREFISH разработана как часть исследования по подбору дозы. В подтверждающей части 2 исследования FIREFISH оценивалась эффективность препарата Эврисди® в терапевтической дозе, которая была выбрана на основании результатов части 1 исследования (см. раздел «Способ применения и дозы»). Пациенты из части 1 не принимали участия в части 2.
В частях 1 и 2 ключевой конечной точкой по эффективности была возможность сидеть без поддержки в течение, как минимум, 5 секунд согласно измерениям по пункту 22 шкалы развития младенцев и детей Бейли – 3 издание (Bayley Scales of Infant and Toddler Development, BSID-III, шкала крупной моторики) после 12 месяцев лечения препаратом Эврисди®.
FIREFISH часть 2
В часть 2 исследования FIREFISH был набран 41 пациент со СМА 1 типа.
Медиана возраста возникновения клинических признаков и симптомов СМА 1 типа составила 1.5 месяцев (1.0-3.0 месяцев), 54% были женского пола, 54% – представители европеоидной расы и 34% – представители азиатской расы.
Медиана возраста на момент набора в исследование составила 5.3 месяцев (2.2-6.9 месяцев), медиана времени между возникновением симптомов и приемом первой дозы составила 3.4 месяцев (1.0-6.0 месяцев).
На исходном уровне средний индекс по результатам теста детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорожденных (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders, CHOP-INTEND) составил 22 балла (8.0-37.0) и средний индекс по результатам неврологической оценки младенцев по шкале Хаммерсмита, модуль 2 (Module 2 of the Hammersmith Infant Neurological Examination, HINE-2) составил 1.0 (0.0-5.0).
Первичной конечной точкой была доля пациентов с возможностью сидеть без поддержки в течение, как минимум, 5 секунд после 12 месяцев терапии (шкала крупной моторики BSID-III, пункт 22). Конечные точки по эффективности у пациентов, получавших препарат Эврисди®, сравнивали с таковыми в аналогичных группах нелеченных пациентов с манифестацией СМА в младенческом возрасте с естественным течением заболевания (критерии функционирования) – см. таблицу 1 ниже.
Таблица 1. Резюме ключевых результатов по эффективности на 12 месяце (FIREFISH часть 2).
| Конечные точки по эффективности | Доля пациентов N=41 (90% ДИ) |
| Основные критерии двигательной функции и развития | |
| BSID-III: способность сидеть без поддержки в течение, как минимум, 5 секунд Значение р на основании критерия функционирования 5%a |
29.3% (17.8%, 43.1%) <0.0001 |
| CHOP-INTEND: индекс 40 или выше Значение р на основании критерия функционирования 17%a |
56.1% (42.1%, 69.4%) <0.0001 |
| CHOP-INTEND: увеличение на ≥4 балла по сравнению с исходным уровнем Значение р на основании критерия функционирования 17%a |
90.2% (79.1%, 96.6%) <0.0001 |
| HINE-2: ответившие по основным критериям двигательной функцииb Значение р на основании критерия функционирования 12%a |
78.0% (64.8%, 88.0%) <0.0001 |
| Выживаемость и выживаемость без событий | |
| Выживаемость без событийc Значение р на основании критерия функционирования 42%a |
85.4% (73.4%, 92.2%) <0.0001 |
| Живы Значение р на основании критерия функционирования 60%a |
92.7% (82.2%, 97.1%) 0.0005 |
| Глотание и кормление | |
| Способность глотать | 85.4% (73.15%, 93.43%) |
| Способность принимать пищу через ротd | 82.9% (70.3%, 91.7%) |
| Использование ресурсов здравоохранения | |
| Отсутствие госпитализацийe | 48.8% (35.1%, 62.6%) |
a Значения р по выживаемости и выживаемости без событий основаны на Z-тестировании; значения р для других конечных точек (BSID-III, CHOP-INTEND, HINE-2) основаны на точном биноминальном тестировании. Доля выживаемости устанавливается с использованием методологии Каплана-Мейера.
b Согласно HINE-2: увеличение на ≥2 балла [или максимальный показатель] для способности брыкаться, ИЛИ увеличение на ≥1 балл по основным критериям двигательной функции: способность держать голову, перекатываться, сидеть, ползать, стоять или ходить И улучшение по большему числу категорий основных критериев двигательной функции по сравнению с ухудшением, что определяется как пациент, ответивший на этот анализ.
c Явлением считается достижение конечной точки по постоянной вентиляции легких (трахеостомия или ≥16 часов неинвазивной вентиляции легких в течение суток или интубация в течение >21 последовательного дня в отсутствие или после разрешения острого обратимого явления). Три пациента достигли конечной точки по постоянной вентиляции легких до 12 месяца. Все 3 пациента достигли увеличения, как минимум, на 4 балла по их индексу CHOP-1NTEND по сравнению с исходным уровнем.
d Включает пациентов, которых кормили исключительно через рот (всего 28 пациентов) и пациентов, которых кормили как через рот, так и с помощью трубки для кормления (всего 6 пациентов) на 12 месяце.
e Госпитализации включают все госпитализации, которые длились, как минимум, 2 дня.
После 12 месяцев терапии препаратом Эврисди® 29% (12/41) пациентов соответствовали критерию способности сидеть без поддержки (BS1D-III, пункт 22), 93% (38/41) пациентов были живы и 85% (35/41) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Эти результаты указывают на клинически значимое отличие от естественного течения заболевания у нелеченных пациентов с манифестацией СМА в младенческом возрасте. Нелеченные пациенты с манифестацией СМА в младенческом возрасте никогда не смогут сидеть без поддержки, и ожидается, что только 25% пациентов смогут выжить без постоянной вентиляции легких после достижения возраста в 14 месяцев.
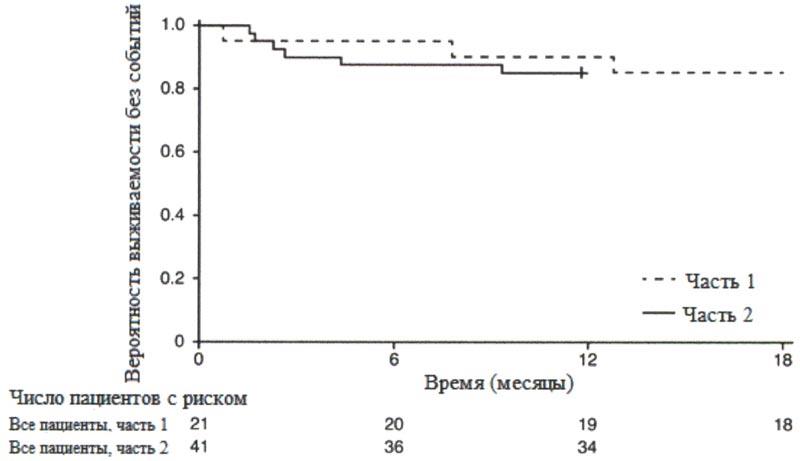 Рисунок 1. График Каплана-Мейера выживаемости без событий (FIREFISH часть 1 и часть 2).
Рисунок 1. График Каплана-Мейера выживаемости без событий (FIREFISH часть 1 и часть 2).
+ цензурировано: один пациент в части 2 был цензурирован, поскольку он рано осуществил визит 12 месяца.
Большинство пациентов достигли ответа по категориям основных критериев двигательной функции HINE-2, включая определенный уровень способности держать голову (76%, 31/41), сидеть (61%, 25/41), перекатываться (56%, 23/41) и стоять (22%, 9/41). Согласно измерениям общего индекса CHOP-INTEND также наблюдались улучшения двигательной функции, см. рисунок 2.
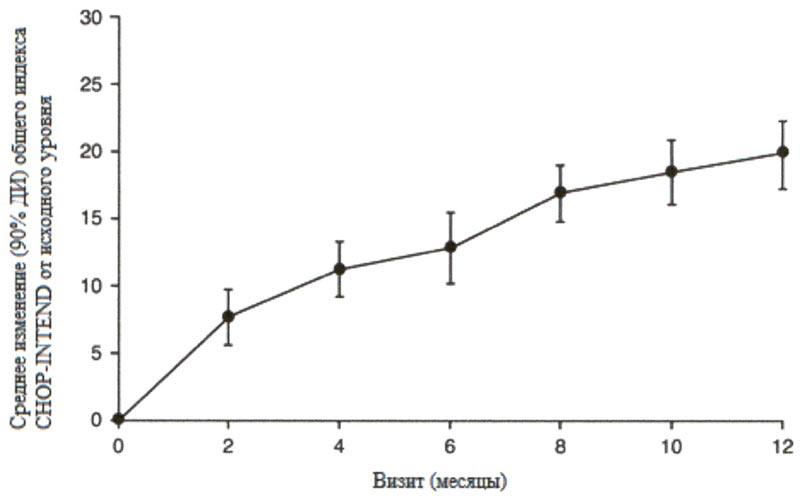 Рисунок 2. Среднее изменение по сравнению с исходным уровнем общего индекса СНОР-INTEND (FIREFISH часть 2).
Рисунок 2. Среднее изменение по сравнению с исходным уровнем общего индекса СНОР-INTEND (FIREFISH часть 2).
FIREFISH часть 1
Эффективность препарата Эврисди® у пациентов со СМА 1 типа также подтверждается результатами из части 1 исследования FIREFISH. Для 21 пациента из части 1 характеристики на исходном уровне согласовывались с таковыми у пациентов с симптомами со СМА 1 типа.
Медиана возраста включения в исследование составила 6.7 месяцев (3.3-6.9 месяцев); медиана времени между возникновением симптомов и приемом первой дозы составила 4.0 месяцев (2.0-5.8 месяцев). Всего 17 пациентов получили терапевтическую дозу (дозу, выбранную для части 2) в течение первых 12 месяцев терапии. После 12 месяцев терапии 41% (7/17) пациентов могли сидеть без поддержки, как минимум, в течение 5 секунд (BSID-III, пункт 22). После 18 месяцев терапии 88% (15/17) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Данные результаты по выживаемости и развитию двигательной функции согласовывались с результатами исследования FIREFISH часть 2.
Поздняя манифестация СМА
ВР39055 (SUNFISH) представляет собой многоцентровое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов со СМА 2 или 3 типа в возрасте 2-25 лет. Исследование состоит из двух частей. В части 1 осуществлялся подбор дозы. Часть 2 представляла собой рандомизированную, двойную слепую, плацебо-контролируемую, подтверждающую часть исследования. Пациенты из части 1 не принимали участия в части 2.
Первичной конечной точкой было изменение оценочного показателя двигательной функции – 32 (Motor Function Measure-32, MFM32) от исходного уровня на 12 месяце. Индекс MFM32 позволяет оценить обширный спектр двигательных функций у большого диапазона пациентов со СМА. Общий индекс MFM32 выражают в виде процента (0-100) от максимально возможного, причем более высокий показатель обозначают лучшую двигательную функцию. Индекс MFM32 измеряет двигательную функцию, в частности важные ежедневные двигательные способности. Небольшие изменения в двигательной функции могут привести к значительному приобретению или потере ежедневных двигательных способностей.
SUNFISH часть 2
SUNFISH часть 2 представляет собой рандомизированную, двойную слепую, плацебо-контролируемую часть исследования SUNFISH у 180 не амбулаторных пациентов со СМА 2 типа (71%) или 3 типа (29%). Пациентов рандомизировали в соотношении 2:1 в группу лечения препаратом Эврисди® в терапевтической дозе (см. раздел «Способ применения и дозы») или в группу плацебо. Рандомизация была стратифицирована по возрастным группам (2-5 лет, 6-11 лет, 12-17 лет и 18-25 лет).
Медиана возраста пациентов на момент начала лечения составляла 9.0 лет (2-25 лет), а медиана времени между возникновением первых симптомов СМА и приемом первой дозы составила 102.6 месяцев (1-275). Из 180 пациентов, включенных в исследование, 51% были женщинами, 67% – представителями европеоидной расы, и 19% – представителями азиатской расы.
На исходном уровне у 67% пациентов выявлен сколиоз (у 32% пациентов – тяжелый сколиоз). Среднее значение индекса MFM32 на исходном уровне составляло 46.1, а индекса по Обновленному модулю оценки функции верхних конечностей (Revised Upper Limb Module, RULM) – 20.1.
В целом, демографические характеристики пациентов на исходном уровне были хорошо сбалансированы между группами Эврисди® и плацебо, за исключением сколиоза (63.3% пациентов в группе препарата Эврисди® в сравнении с 73.3% пациентов в группе плацебо).
Первичный анализ изменения общего индекса MFM32 на 12 месяце по сравнению с исходным уровнем показал клинически и статистически значимое различие между пациентами, получавшими терапию препаратом Эврисди®, и пациентами, получавшими плацебо, в исследовании SUNFISH часть 2. Результаты первичного анализа и ключевых вторичных конечных точек представлены в таблице 2 и на рисунках 3 и 4.
Таблица 2. Резюме данных по эффективности у пациентов с поздней манифестацией СМА на 12 месяце лечения (SUNFISF1 часть 2).
| Конечная точка | Эврисди® (N=120) |
Плацебо (N=60) |
| Первичная конечная точка | ||
| Изменение общего индекса MFM321 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95% ДИ) |
1.36 (0.61,2.11) |
-0.19 (-1.22, 0.84) |
| Различие по сравнению с плацебо Расчетное значение (95% ДИ) Значение р2 |
1.55 (0.30, 2.81) 0.0156 |
|
| Вторичные конечные точки | ||
| Доля пациентов с изменением общего индекса MFM321 на 12 месяце на ≥3 балла от исходного уровня (95% ДИ) | 38.3% (28.9, 47.6) |
23.7% (12.0, 35.4) |
| Отношение шансов для общего ответа (95% ДИ) Значение р3,4 с коррекцией (без коррекции) |
2.35 (1.01,5.44) 0.0469 (0.0469) |
|
| Изменение общего индекса RULM5 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95% ДИ) |
1.61 (1.00, 2.22) |
0.02 (-0.83, 0.87) |
| Различие по сравнению с плацебо Расчетное (95% ДИ) значение р2,4 с коррекцией (без коррекции) |
1.59(0.55, 2.62) 0.0469 (0.0028) |
НК = наименьшие квадраты
1 На основании правила по отсутствующим данным для индекса MFM32 6 пациентов были исключены из анализа (Эврисди® n = 115, контрольная группа плацебо n = 59).
2 Данные анализировали с использованием смешанной модели повторного измерения с общим индексом на исходном уровне, лечением, визитом, возрастной группой, лечением к визиту, а также исходным уровнем к визиту.
3 Данные анализировали с использованием логистической регрессии с общим индексом на исходном уровне, лечением и возрастной группой.
4 Значение р с коррекцией было получено для конечных точек, включенных в иерархическое тестирование, и выделялось на основании всех значений р, полученных для конечных точек в порядке увеличения иерархии до текущей конечной точки. Значение р без коррекции тестировали при уровне значимости 5%.
5 На основании правила отсутствующих данных для индекса RULM из анализа исключили 3 пациентов (Эврисди® n = 119; контрольная группа плацебо, n = 58).
При сравнении с группой плацебо пациенты, получавшие лечение препаратом Эврисди®, продемонстрировали значительное улучшение двигательной функции согласно оценке индекса MFM32 (среднее различие составило 1.55 баллов; р=0.0156) после 12 месяцев лечения. Пациенты в возрасте 2-5 лет, получавшие терапию препаратом Эврисди®, продемонстрировали наибольшее улучшение индекса MFM32 по сравнению с контрольной группой плацебо (увеличение на ≥3 балла у 78.1% в сравнении с 52.9%). Пациенты ≥18 лет, получавшие терапию препаратом Эврисди®, достигли стабилизации заболевания (изменение от исходного уровня по общему индексу MFM32 составило ≥0 баллов: 57.1% в сравнении с 37.5%).
У пациентов со СМА 2 типа и 3 типа, получавших терапию препаратом Эврисди®, наблюдалось сопоставимое улучшение по сравнению с исходным уровнем согласно индексу MFM32 (1.54 баллов [95% ДИ: 0.06, 3.02]; 1.49 баллов [95% ДИ: -0.94, 3.93] соответственно) по сравнению с контрольной группой плацебо.
Исследование также достигло вторичного независимого исхода двигательной функции, RULM. Согласно RULM отмечались статистически и клинически значимые улучшения в двигательной функции после 12 месяцев лечения по сравнению с исходным уровнем. Пациенты в возрасте 2-5 лет, получавшие лечение препаратом Эврисди®, продемонстрировали наибольшее улучшение индекса RULM (3.41 баллов [95% ДИ: 1.55, 5.26]), улучшение также отмечалось у пациентов ≥18 лет (1.74 баллов [95% ДИ: -1.06, 4.53]).
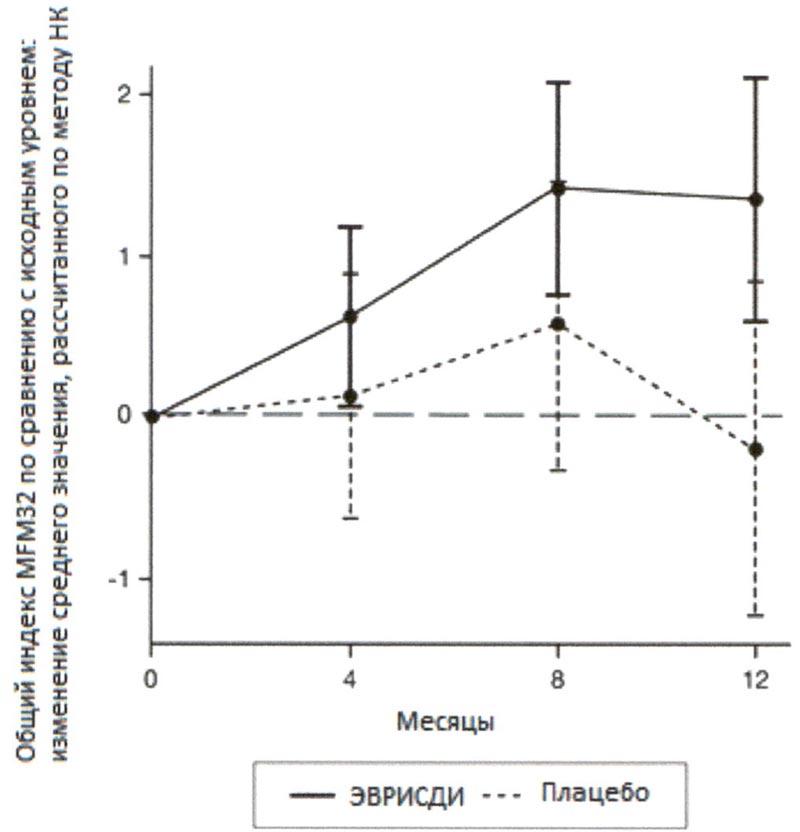 Рисунок 3. Изменение среднего значения общего индекса MFM32 в течение 12 месяцев от исходного уровня (исследование SUNFISH часть 2)1.
Рисунок 3. Изменение среднего значения общего индекса MFM32 в течение 12 месяцев от исходного уровня (исследование SUNFISH часть 2)1.
1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу MFM32 [95% ДИ].
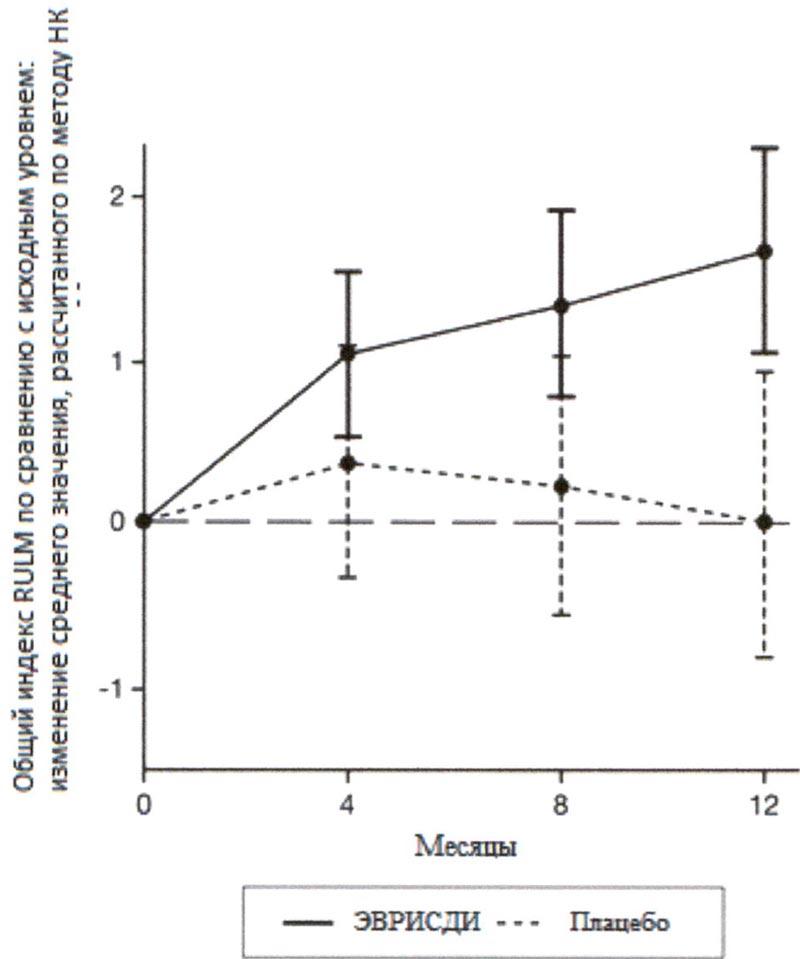 Рисунок 4. Изменение среднего значения общего индекса RULM в течение 12 месяцев от исходного уровня (SUNFISH часть 2)1.
Рисунок 4. Изменение среднего значения общего индекса RULM в течение 12 месяцев от исходного уровня (SUNFISH часть 2)1.
1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу RULM [95% ДИ].
SUNFISH часть 1
Эффективность препарата Эврисди® у пациентов с поздней манифестацией СМА также подтверждается результатами из части 1 (часть исследования SUNFISH по подбору дозы). В часть 1 был включен 51 пациент со СМА 2 типа и 3 типа (включая 7 амбулаторных пациентов) в возрасте 2-25 лет.
После 1 года лечения препаратом в терапевтической дозе (доза, выбранная для части 2) было отмечено клинически значимое улучшение двигательной функции согласно измерениям индекса MFM32. Среднее изменение по сравнению с исходным уровнем составило 2.7 баллов (95% ДИ: 1.5, 3.8). Улучшение индекса MFM32 сохранялось ≤2 лет во время лечения препаратом Эврисди® (среднее изменение на 2.7 баллов [95% ДИ: 1.2, 4.2]).
В поисковом анализе двигательную функцию, оцениваемую согласно индексу MFM32, в части 1 исследования SUNFISH сравнивали с группой естественного течения СМА (на основании ключевых прогностических факторов). Общее изменение индекса MFM по сравнению с исходным уровнем после 1 года и 2 лет было выше у пациентов, получавших препарат Эврисди®, по сравнению с группой естественного течения заболевания (после 1 года: различие на 2.7 баллов; р <0.0001; после 2 лет: различие на 4.0 балла; р <0.0001). В группе естественного течения заболевания отмечалось ухудшение двигательной функции, как и ожидалось для естественного прогрессирования СМА (после 1 года: среднее изменение -0.6; после 2 лет: среднее изменение -2.0).
Применение у пациентов, ранее получавших лечение по поводу СМА
ВР39054 (JEWELFISH) представляет собой одногрупповое, открытое исследование по изучению безопасности, переносимости, фармакокинетики и фармакодинамики препарата Эврисди® у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. В исследование были включены пациенты в возрасте 6 месяцев – 60 лет, которые ранее получали лечение по поводу СМА (включая нусинерсен и онасемноген абепарвовек). Из 174 пациентов, включенных в исследование, 76 пациентов ранее получали терапию нусинерсеном (9 пациентов со СМА 1 типа, 43 пациента со СМА 2 типа и 24 пациента со СМА 3 типа) и 14 пациентов ранее получали онасемноген абепарвовек (4 пациента со СМА 1 типа и 10 пациентов со СМА 2 типа). После 4 недель терапии препаратом Эврисди® у пациентов отмечалось увеличение уровня белка SMN в крови в среднем ≥2 раза выше по сравнению с исходным уровнем.
Фармакокинетика
Фармакокинетические параметры препарата Эврисди® были изучены у здоровых взрослых добровольцев и у пациентов со СМА.
После приема препарата Эврисди® в виде раствора внутрь фармакокинетика рисдиплама была приблизительно линейной между 0.6 и 18 мг. Фармакокинетика рисдиплама наилучшим образом описывалась с помощью популяционной фармакокинетической модели со всасыванием с тремя транзитными камерами, двухкамерным распределением и выведением первого порядка. Было обнаружено, что масса тела и возраст оказывают значимое влияние на фармакокинетику.
При применении рисдиплама в терапевтической дозе 0.2 мг/кг один раз в сутки у пациентов с манифестацией СМА в младенческом возрасте (от 2 до 7 месяцев при наборе в исследование) расчетная экспозиция (средняя площадь под кривой «концентрация-время» (AUC)0-24) составила 1930 нг.ч/мл. В исследовании SUNFISH (часть 2) при применении рисдиплама в терапевтической дозе 0.25 мг/кг один раз в сутки у пациентов с массой тела <20 кг и 5 мг один раз в сутки у пациентов с массой тела ≥20 кг (пациенты с поздней манифестацией СМА от 2 до 25 лет при наборе в исследование), расчетная экспозиция составила 2070 нг.ч/мл. Наблюдаемая максимальная концентрация (средняя Сmax) составила 194 нг.ч/мл при применении в дозе 0.2 мг/кг в исследовании F1REFISH и 120 нг.ч/мл в исследовании SUNFISH часть 2.
Всасывание
Рисдиплам быстро всасывался при приеме натощак, при этом время достижения максимальной концентрации (tmax) в плазме варьировало от 1 до 4 часов после приема внутрь. Прием пищи (высококалорийный завтрак с высоким содержанием жиров) не оказывал значимого влияния на экспозицию рисдиплама.
Распределение
Установленные популяционные фармакокинетические параметры составили 98 л для кажущегося центрального объема распределения, 93 л для периферического объема и 0.68 л/ч для межкамерного клиренса.
Рисдиплам преимущественно связывался с сывороточным альбумином без какого-либо связывания с альфа-1-кислым гликопротеином, свободная фракция составила 11%.
Метаболизм
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1А1, 2J2, 3А4 и 3А7.
Одновременный прием итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приемом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11%, показатель Сmax снижен на 9%).
Выведение
В ходе популяционного фармакокинетического анализа был установлен кажущийся клиренс (CL/F) рисдиплама 2.6 л/ч.
Эффективный период полувыведения рисдиплама составил приблизительно 50 часов у пациентов со СМА.
Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Приблизительно 53% дозы (14% неизменного рисдиплама) выводилось с калом и 28% с мочой (8% неизменного рисдиплама). Исходный препарат был основным компонентом в плазме, что составило 83% от общих компонентов препарата, находящихся в кровотоке. Фармакологически неактивный метаболит М1 был определен как основной циркулирующий метаболит.
Особые группы пациентов
Пациенты детского возраста
Масса тела и возраст были определены как ковариаты в популяционном фармакокинетическом анализе. Таким образом, доза определяется в зависимости от возраста (младше и старше 2 лет) и массы тела (до 20 кг) для достижения сходных экспозиций среди пациентов всех возрастов и с различной массой тела. Данные у пациентов младше 2 месяцев отсутствуют.
Пациенты пожилого возраста
Специальных исследований по изучению фармакокинетики препарата Эврисди® у пациентов со СМА старше 60 лет не проводилось. Пациенты со СМА ≤60 лет принимали участие в исследовании JEWELFISH. Субъекты без СМА ≤69 лет принимали участие в клинических исследованиях по фармакокинетике. Согласно результатам данных исследований коррекции дозы у пациентов ≤69 лет не требуется.
Пациенты с нарушением функции почек
Исследований по изучению фармакокинетики рисдиплама у пациентов с нарушением функции почек не проводилось. Рисдиплам в виде неизменного соединения выводится почками в малой степени (8%).
Пациенты с нарушением функции печени
Нарушение функции печени легкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. После применения 5 мг рисдиплама средние соотношения для Сmax и AUC составили 0.95 и 0.80 у субъектов с нарушением функции печени легкой степени тяжести (n=8), а также 1.20 и 1.08 у субъектов с нарушением функции печени средней степени тяжести (п=8) по сравнению с соответствующими здоровыми добровольцами (n=10). Безопасность и фармакокинетика у пациентов с нарушением функции печени тяжелой степени тяжести не изучались.
Этническая принадлежность
Фармакокинетика рисдиплама у японских субъектов и субъектов европеоидной расы не отличалась.
Показания к применению
Лечение спинальной мышечной атрофии (СМА) у взрослых и детей с 2 месяцев.
Противопоказания
Повышенная чувствительность к рисдипламу или к другим вспомогательным веществам препарата в анамнезе.
Беременность и период грудного вскармливания.
Младенцы младше 2 месяцев.
Применение при беременности и в период грудного вскармливания
Беременность
Применение препарата Эврисди® при беременности противопоказано.
Данные клинических исследований препарата Эврисди® у беременных женщин отсутствуют. Была показана эмбриофетальная токсичность и тератогенность рисдиплама у животных. На основании данных, полученных в исследованиях у животных, установлено, что рисдиплам проходит через плацентарный барьер и может оказывать повреждающее действие на плод.
Безопасность применения препарата Эврисди® во время родов и родоразрешения не установлена.
Период грудного вскармливания
Применение препарата в период грудного вскармливания противопоказано. Неизвестно, выводится ли препарат Эврисди® с грудным молоком у человека. Исследования у крыс показали, что рисдиплам выводится с грудным молоком.
Фертильность
Пациенты-мужчины
Согласно данным доклинических исследований фертильность у мужчин может быть нарушена в ходе терапии препаратом Эврисди®. В репродуктивных органах крыс и обезьян наблюдались дегенеративные формы и снижение числа сперматозоидов. Влияние на сперматозоиды обратимо после отмены рисдиплама.
Перед началом лечения препаратом Эврисди® с пациентами-мужчинами необходимо обсудить стратегии сохранения фертильности. Пациенты-мужчины могут рассмотреть возможность консервации спермы перед началом лечения или по прошествии минимум 4 месяцев после завершения лечения. Пациенты-мужчины, которые хотят завести ребенка, должны прекратить лечение препаратом Эврисди®, как минимум, на 4 месяца. Лечение может быть продолжено после зачатия.
Пациенты-женщины
На основании данных доклинических исследований влияния препарата Эврисди® на фертильность у женщин не ожидается.
Тестирование на беременность
Перед началом терапии препаратом Эврисди® следует проверить статус беременности у женщин репродуктивного потенциала. Беременных женщин следует четко проинформировать о потенциальном риске для плода.
Контрацепция
Пациенты (мужчины и женщины) репродуктивного потенциала должны соблюдать следующие требования в отношении контрацепции:
- пациенты-женщины детородного потенциала должны использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приема последней дозы;
- пациенты-мужчины и их партнерши детородного потенциала должны вместе использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 4 месяцев после приема последней дозы пациентом-мужчиной.
Способ применения и дозы
Раствор для приема внутрь должен быть приготовлен медицинским работником перед его выдачей пациенту.
Общие рекомендации
Лечение СМА следует начать как можно раньше после постановки диагноза.
Препарат Эврисди® принимают внутрь один раз в сутки приблизительно в одно и то же время каждый день с помощью предоставляемого перорального шприца.
Рекомендуемая суточная доза препарата Эврисди® у пациентов со СМА определяется в зависимости от возраста и массы тела (см. таблицу 3).
Таблица 3. Режим дозирования в зависимости от возраста и массы тела.
| Возраст и масса тела | Рекомендуемая суточная доза |
| от 2 месяцев до <2 лет | 0.20 мг/кг |
| ≥2 лет (масса тела <20 кг) | 0.25 мг/кг |
| ≥2 лет (масса тела ≥20 кг) | 5 мг |
Изменения дозы должны проводиться под контролем медицинского работника. Терапия препаратом в дозах выше 5 мг в сутки не изучалась. Данные у младенцев младше 2 месяцев отсутствуют.
Способ применения
Для приема суточной дозы препарата Эврисди® следует использовать предоставляемый многоразовый пероральный шприц. Медицинский работник перед началом лечения должен обсудить с пациентом или лицом, осуществляющим уход за пациентом, как подготовить назначенную суточную дозу для приема.
Пациенту необходимо выпить воду после приема препарата Эврисди® для гарантии, что препарат проглочен полностью. В случае, если пациент не может глотать и у него установлена назогастральная или гастростомическая трубка, следует ввести препарат через трубку. После введения препарата Эврисди® необходимо промыть трубку водой.
Задержка введения или пропуск дозы
Препарат Эврисди® принимают внутрь один раз в сутки приблизительно в одно и то же время каждый день.
Если прием препарата Эврисди® был пропущен и прошло не более 6 часов с момента запланированного приема, следует принять препарат как можно скорее.
Если прошло более 6 часов с момента запланированного приема, следует пропустить прием и принять препарат в обычное запланированное время на следующий день.
Если препарат не был проглочен полностью или после его приема произошла рвота, не следует принимать препарат повторно для восполнения неполной дозы. Необходимо дождаться следующего дня и принять препарат в обычное запланированное время.
Дозирование в особых случаях
Пациенты детского возраста
Эффективность и безопасность препарата Эврисди® у пациентов <2 месяцев еще не установлена (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Пациенты пожилого возраста
Фармакокинетика и безопасность препарата Эврисди® оценивались у субъектов без СМА в возрасте ≤69 лет. Применение препарата Эврисди® у пациентов со СМА старше 60 лет не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов»).
Пациенты с нарушением функции почек
Эффективность и безопасность препарата Эврисди® у пациентов с нарушением функции почек не изучались. Не ожидается необходимости в коррекции дозы у пациентов с нарушением функции почек (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов»).
Пациенты с нарушением функции печени
Коррекции дозы у пациентов с нарушением функции печени легкой или средней степени тяжести не требуется. Применение препарата Эврисди® у пациентов с нарушением функции печени тяжелой степени тяжести не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов» и раздел «Особые указания»).
Особая информация об использовании, обращении и утилизации препарата
Приготовление раствора препарата Эврисди® из порошка осуществляется медицинским работником перед выдачей пациенту.
Приготовление раствора для приема внутрь 0.75 мг/мл из 60 мг порошка (см. в конце инструкции Руководство по разведению – только для медицинских работников).
Следует соблюдать осторожность при обращении с порошком препарата Эврисди® для приготовления раствора для приема внутрь (см. раздел «Особые указания»). Следует избегать вдыхания и прямого контакта сухого порошка и приготовленного раствора с кожей или слизистыми оболочками. Следует использовать одноразовые перчатки во время разведения и при очистке наружной поверхности флакона/крышки и рабочей поверхности после разведения.
Если контакт все же произошел, нужно тщательно промыть данный участок водой с мылом, а глаза – просто водой.
Выбор перорального шприца для применения назначенной суточной дозы препарата Эврисди®
Таблица 4. Выбор перорального шприца для применения назначенной суточной дозы.
| Размер шприца | Объем дозы | Деления на шприце |
| 6 мл | 1.0 мл-6.0 мл | 0.1 мл |
| 12 мл | 6.2 мл-6.6 мл | 0.2 мл |
При расчете объема дозы следует принять во внимание деления на шприце. Необходимо округлить объем дозы до ближайшего деления, отмеченного на выбранном пероральном шприце.
Пациенты должны принять препарат Эврисди® незамедлительно после его набора в пероральный шприц. Если препарат не был принят в течение 5 минут, следует утилизировать раствор препарата из перорального шприца и набрать новую дозу.
Несовместимость
Не обнаружено признаков несовместимости между препаратом Эврисди® и рекомендуемыми пероральными шприцами.
Инструкции по уничтожению неиспользованного препарата или препарата с истекшим сроком годности
Попадание лекарственного препарата в окружающую среду должно быть сведено к минимуму. Не следует утилизировать препарат с помощью сточных вод или вместе с бытовыми отходами. Уничтожение неиспользованного препарата или препарата с истекшим сроком годности должно проводиться в соответствии с локальными требованиями.
Руководство по использованию препарата
Пациенты/лица, осуществляющие уход за пациентами, должны ознакомиться с важной информацией касательно приема препарата, которая представлена ниже.
Важная информация для пациентов/лиц, осуществляющих уход за пациентами, по препарату Эврисди®
- Попросите медицинского работника показать Вам правильный шприц, который Вы должны использовать, и как отмерять назначенную суточную дозу.
- Всегда используйте многоразовые пероральные шприцы, предоставляемые в упаковке, для того, чтобы отмерять назначенную Вам суточную дозу. Пероральные шприцы защищают препарат от света.
- Предоставляется по два пероральных шприца каждого размера на случай, если один из них будет утерян или поврежден. Если оба пероральных шприца утеряны или повреждены, свяжитесь с медицинским работником, который проконсультирует Вас, как можно продолжить применение препарата.
- См. выше подраздел «Выбор перорального шприца для применения назначенной суточной дозы», чтобы выбрать правильный пероральный шприц для использования. Проконсультируйтесь с медицинским работником в случае возникновения вопросов по выбору правильного перорального шприца.
- Не применяйте препарат Эврисди®, если во флаконе отсутствует адаптер для флакона, и свяжитесь с медицинским работником.
- Не применяйте препарат Эврисди® последаты утилизации, указанной на этикетке флакона. Если на этикетке флакона отсутствует дата утилизации, свяжитесь с медицинским работником.
- Не смешивайте препарат Эврисди® с пищей и напитками.
- Не используйте препарат Эврисди®, если флакон или пероральные шприцы повреждены.
- Избегайте контакта препарата Эврисди® с кожей. Если это произошло, промойте данный участок водой с мылом.
- Если Вы разлили препарат Эврисди®, протрите данную область сухим бумажным полотенцем и промойте водой с мылом. Бумажное полотенце следует выбросить в мусор и хорошо вымыть руки водой с мылом.
- Если во флаконе осталось количество препарата, недостаточное для приема назначенной дозы, утилизируйте флакон с оставшимся препаратом и использованные пероральные шприцы в соответствии с локальными требованиями; используйте новый флакон с препаратом Эврисди® для приема назначенной дозы. Не смешивайте препарат Эврисди® из нового флакона с препаратом Эврисди® из используемого на данный момент флакона.
Хранение раствора препарата Эврисди®
Следует хранить приготовленный раствор препарата в оригинальном флаконе темного стекла, плотно закрытом крышкой, в холодильнике (при температуре 2-8 °С) в вертикальном положении, в течение до 64 дней после разведения. Не замораживать.
А) Подготовка и отбор назначенной дозы
Как выбрать правильный пероральный шприц для применения назначенной дозы препарата Эврисди®
Если назначенный объем суточной дозы составляет от 1 мл до 6 мл, следует использовать пероральный шприц на 6 мл (с серой этикеткой).
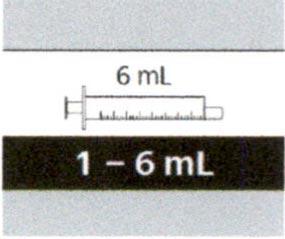
Если назначенный объем суточной дозы составляет от 6.2 мл и выше, следует использовать пероральный шприц на 12 мл (с коричневой этикеткой).

Как подготовить назначенный объем дозы препарата Эврисди®
Шаг А1
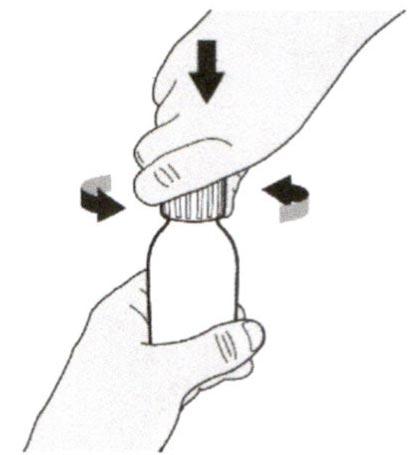
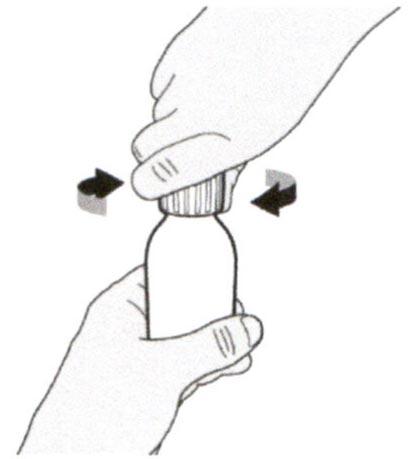
Снимите крышку, нажав на нее вниз и затем повернув налево (против часовой стрелки). Не выбрасывайте крышку.
Шаг А2
Нажимайте на поршень перорального шприца до конца для удаления воздуха.
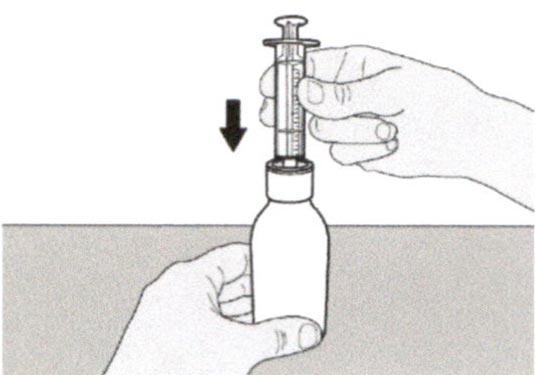
Шаг А3
Держа флакон вертикально, вставьте кончик шприца в адаптер для флакона.
Шаг А4
Аккуратно переверните флакон вместе с пероральным шприцем, кончик которого плотно вставлен в адаптер для флакона.
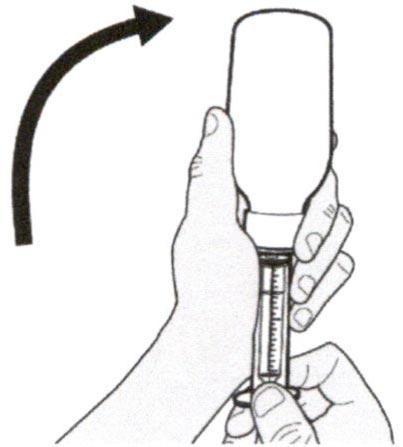
Шаг А5
Медленно оттягивайте поршень, чтобы набрать назначенный объем дозы препарата Эврисди®.
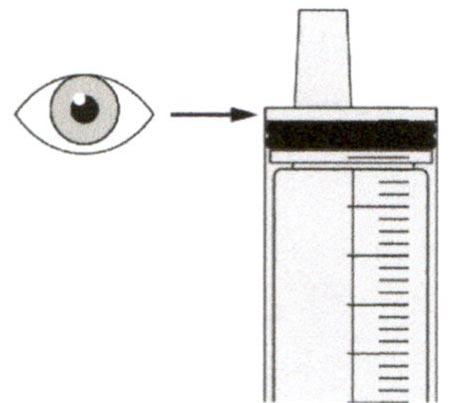
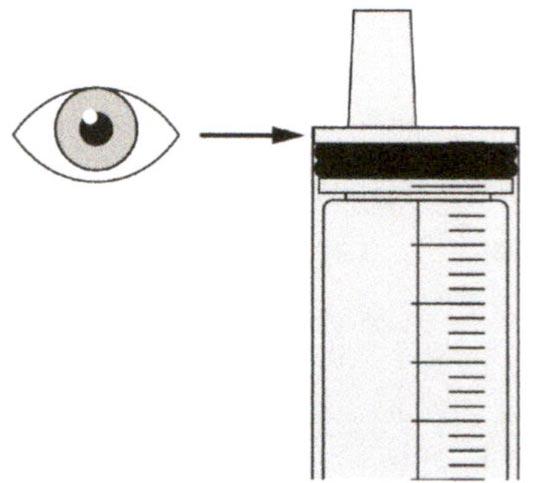
Верхняя поверхность черного ограничителя поршня должна находиться на одной линии с делением (мл) на пероральном шприце, соответствующем назначенному объему суточной дозы.
После того, как доза была набрана правильно, удерживайте поршень на месте и не давайте ему смещаться.
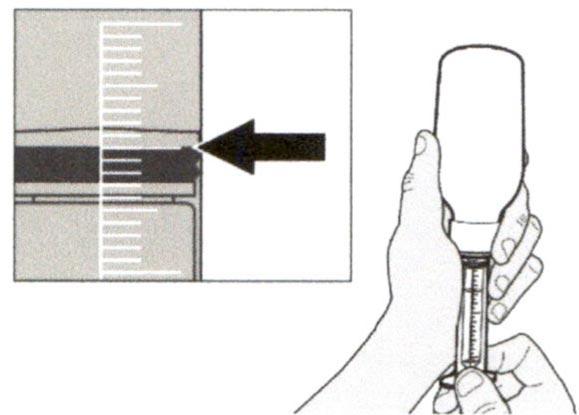
Шаг А6
Продолжайте удерживать поршень на месте и не давайте ему смещаться. Оставьте пероральный шприц в адаптере для флакона и поверните флакон вертикально, горлышком вверх. Поместите флакон на плоскую поверхность. Удалите пероральный шприц из адаптера для флакона, аккуратно вытягивая прямо вверх пероральный шприц.
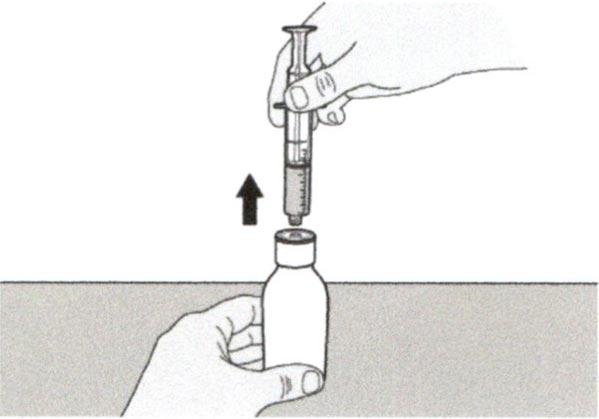
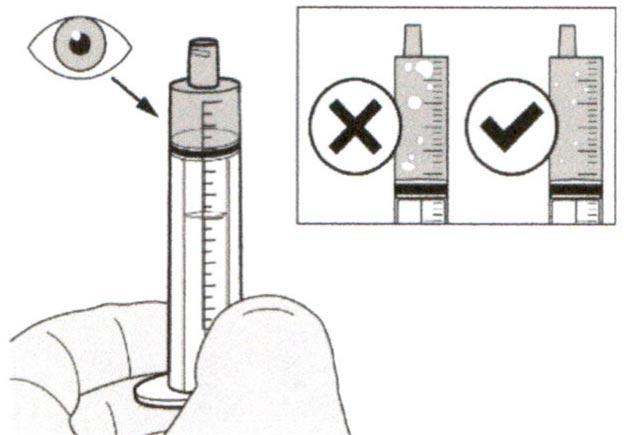
Шаг А7
Удерживайте пероральный шприц, чтобы его кончик был направлен вверх. Проверьте препарат в пероральном шприце. Если в растворе присутствуют большие пузырьки воздуха или если Вы неправильно набрали дозу препарата Эврисди®, плотно вставьте кончик шприца в адаптер для флакона. Нажимайте на поршень шприца до конца, чтобы переместить препарат обратно во флакон и повторите шаги А4-А7.
Примите или введите препарат Эврисди® незамедлительно после его набора в пероральный шприц.
Если препарат не был принят в течение 5 минут, следует утилизировать раствор препарата из перорального шприца и набрать новую дозу.
Шаг А8
Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы плотно закрыть флакон.
Не удаляйте адаптер для флакона из флакона.
Если Вы принимаете препарат Эврисди® перорально, см. пункт Б)
Прием препарата Эврисди® перорально.
Если Вы вводите препарат Эврисди® через гастростомическую трубку, см. пункт В)
Введение препарата Эврисди® через гастростомическую трубку.
Если Вы вводите препарат Эврисди® через назогастральную трубку, см. пункт Г)
Введение препарата Эврисди® через назогастральную трубку.
Б) Прием препарата Эврисди® перорально
Следует сесть прямо для перорального приема препарата Эврисди®.
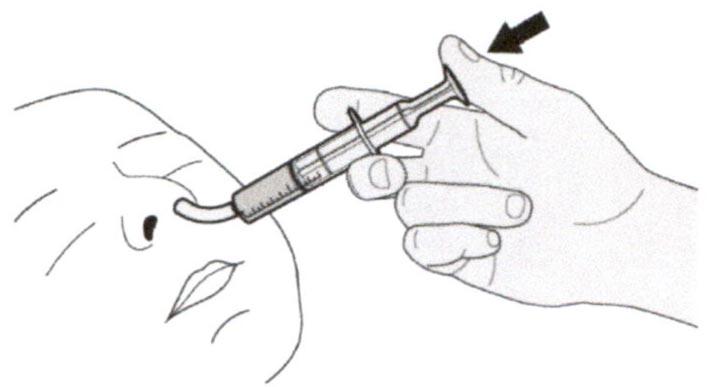
Шаг Б1
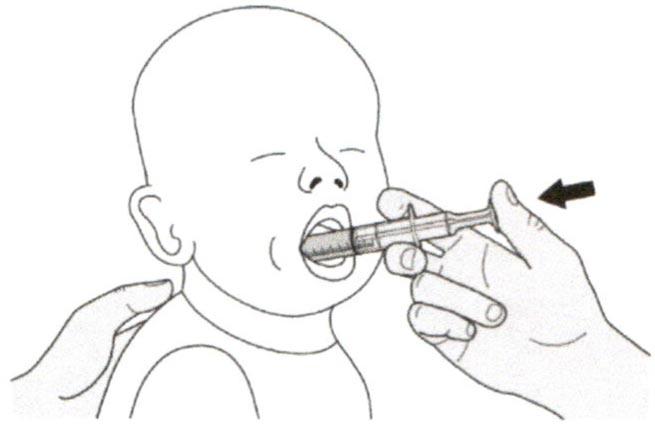
Поместите пероральный шприц в рот, расположив кончик шприца вдоль любой щеки.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
Введение препарата Эврисди® прямо в горло или слишком быстро может привести к удушью.
Шаг Б2
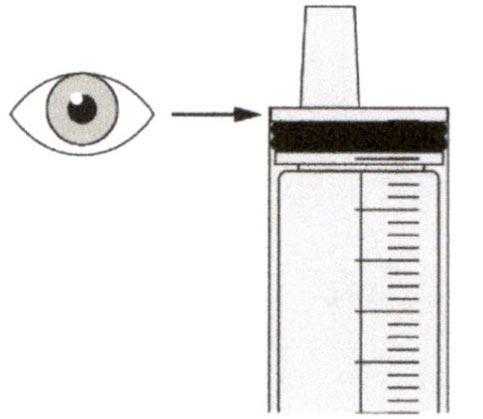
Проверьте, что в пероральном шприце не остался раствор препарата.
Шаг Б3
Выпейте воду сразу после приема назначенной дозы препарата Эврисди®.
Затем см. Шаг Д по очистке шприца.
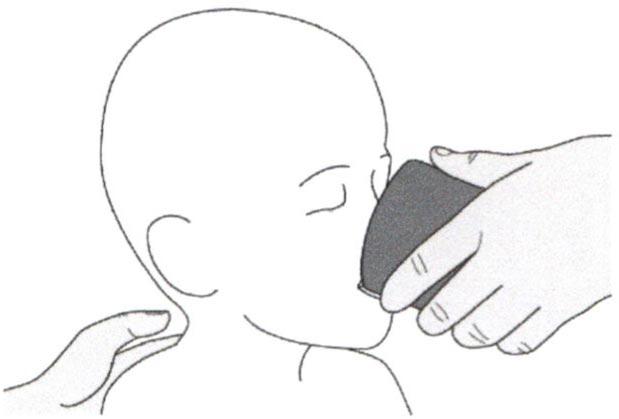
В) Введение препарата Эврисди® через гастростомическую трубку
Если Вы вводите препарат Эврисди® через гастростомическую трубку, проконсультируйтесь с медицинским работником касательно проверки гастростомической трубки перед введением препарата Эврисди®.
Шаг В1
Поместите кончик перорального шприца в гастростомическую трубку.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
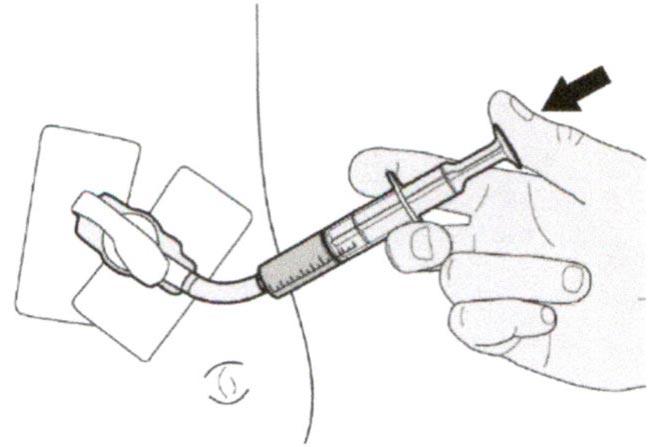
Шаг В2
Проверьте, что в пероральном шприце не остался раствор препарата.
Шаг В3
Сразу после введения назначенной дозы препарата Эврисди® следует промыть гастростомическую трубку 10-20 мл воды.
Затем см. Шаг Д по очистке шприца.
Г) Введение препарата Эврисди® через назогастральную трубку
Если Вы вводите препарат Эврисди® через назогастральную трубку, проконсультируйтесь с медицинским работником касательно проверки назогастральной трубки перед введением препарата Эврисди®.
Шаг Г1
Поместите кончик перорального шприца в назогастральную трубку.
Медленно нажимайте на поршень до конца для введения полной дозы препарата Эврисди®.
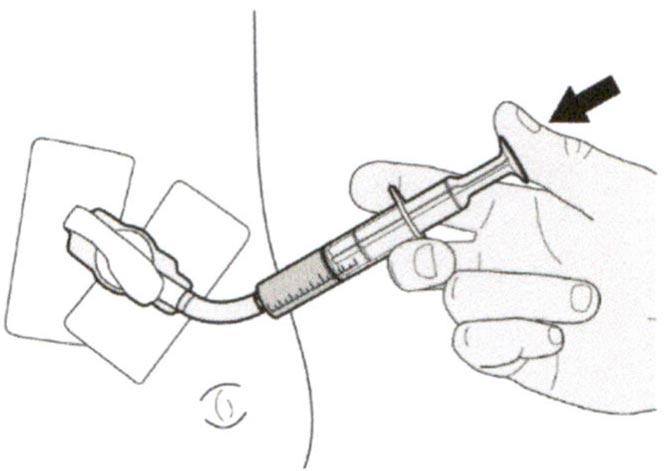
Шаг Г2
Проверьте, что в пероральном шприце не остался раствор препарата.
Шаг Г3
Сразу после введения назначенной дозы препарата Эврисди® следует промыть назогастральную трубку 10-20 мл воды.
Затем см. Шаг Д по очистке шприца.
Д) Очистка перорального шприца после использования
Шаг Д1
Удалите поршень из перорального шприца.
Хорошо промойте цилиндр перорального шприца чистой водой.
Шаг Д2
Хорошо промойте поршень перорального шприца чистой водой.
Шаг Д3
Убедитесь, что цилиндр и поршень перорального шприца чистые.
Поместите цилиндр и поршень перорального шприца на чистую поверхность в безопасном месте, чтобы они высохли.
Вымойте руки.
После того как цилиндр и поршень перорального шприца высохнут, вставьте поршень в цилиндр перорального шприца и храните вместе с препаратом для дальнейшего использования.
Побочное действие
Резюме профиля безопасности
Профиль безопасности препарата Эврисди® основан на трех клинических исследованиях FIREFISH, SUNFISH и JEWELFISH.
FIREFISH представляет собой открытое исследование, состоящее из двух частей, в которое включено 62 пациента с манифестацией СМА в младенческом возрасте (от 2.2 до 6.9 месяцев). Пятьдесят пять пациентов получали терапию препаратом Эврисди® в течение >12 месяцев (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»). Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с манифестацией СМА в младенческом возрасте, представленные в таблице 5 ниже, основаны на данных объединенного анализа у пациентов из частей 1 и 2 клинического исследования FIREFISH. Нежелательные реакции определяются как нежелательные явления, которые отмечаются у ≥5% пациентов и для которых возможна причинно-следственная связь с препаратом Эврисди®.
SUNFISH представляет собой исследование, состоящее из двух частей, в которое включены пациенты с поздней манифестацией СМА (от 2 до 25 лет) (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с поздней манифестацией СМА, основаны на данных исследования SUNFISH часть 2 (n=180), рандомизированной, двойной слепой, плацебо-контролируемой части исследования с периодом последующего наблюдения, как минимум, 12 месяцев. Нежелательные реакции определяются как нежелательные явления, которые отмечаются на ≥5% чаще или, как минимум, в 2 раза чаще по сравнению с пациентами, получавшими плацебо, и для которых возможна причинно-следственная связь с препаратом Эврисди®.
Таблица 5. Резюме нежелательных реакций у пациентов с манифестацией СМ А в младенческом возрасте в исследовании FIREFISH (части 1 и 2).
| Класс систем органов | Нежелательная реакция |
Частота N=62 n (%) |
Число явлений/ 100 пациенто-лет Общая экспозиция пациенто-лет = 87.9 | Категория частоты |
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 10 (16.1) | 13.7 | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 17 (27.4) | 23.9 | Очень часто |
* Включая сыпь, макулопапулезную сыпь, эритему, дерматит, аллергический дерматит, папулезную сыпь, фолликулит.
Таблица 6. Резюме нежелательных реакций у пациентов с поздней манифестацией СМА в исследовании SUNFISH часть 2.
| Класс систем органов | Нежелательная реакция | Частота N=120 n (%) |
Плацебо N=60 n (%) |
Категория частоты |
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 20 (16.7) | 5 (8.3%) | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 20 (16.7) | 1 (1.7) | Очень часто |
* Включая сыпь, макулопапулезную сыпь, эритему, аллергический дерматит, эритематозную сыпь, фолликулит, папулезную сыпь.
Нежелательные реакции диарея и сыпь отмечались без определенного времени или клинической картины и разрешались несмотря на продолжающееся лечение препаратом Эврисди® у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. Данные явления не указывают на влияние на эпителиальные ткани, которые отмечались в исследованиях у животных.
Профиль безопасности у пациентов, ранее получавших терапию по поводу СМА
Профиль безопасности препарата Эврисди® у пациентов, ранее получавших терапию по поводу СМА в исследовании JEWELFISH, сопоставим с таковым у пациентов, ранее не получавших лечения в исследованиях FIREFISH (часть 1 и 2) и SUNFISH (часть 1 и 2). В исследовании JEWELFISH принимали участие 76 пациентов, которые ранее получали нусинерсен и 14 пациентов, которые ранее получали онасемноген абепарвовек (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Передозировка
Случаев передозировки препарата Эврисди® в клинических исследованиях не было.
Лечение
Известного антидота для применения в случае передозировки препарата Эврисди® нет.
В случае передозировки следует тщательно наблюдать за пациентом и провести поддерживающую терапию.
Взаимодействие с другими лекарственными средствами
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1А1, 2J2, ЗА4 и ЗА7. Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Влияние других лекарственных средств на препарат Эврисди®
Одновременный прием итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приемом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11%, показатель Сmax снижен на 9%). При одновременном применении ингибитора CYP3A и препарата Эврисди® коррекции дозы последнего не требуется.
Лекарственного взаимодействия с препаратами, метаболизирующимися посредством флавин-монооксигеназы 1 и 3 (FMO 1 и 3), не ожидается.
Влияние препарата Эврисди® на другие лекарственные средства
В условиях in vitro рисдиплам и его основной циркулирующий метаболит М1 не индуцировали CYP1A2, 2В6, 2С8, 2С9, 2С19 или 3А4. В условиях in vitro рисдиплам и M1 не ингибировали (обратимо или в зависимости от времени) какой-либо из изучаемых ферментов CYP (CYP1A2, 2В6, 2С8, 2С9, 2С19, 2D6) за исключением CYP3A.
Препарат Эврисди® является слабым ингибитором CYP3A. При введении препарата Эврисди® здоровым взрослым добровольцам один раз в сутки в течение двух недель экспозиция мидазолама (чувствительного субстрата CYP3A) незначительно повышалась (AUC 11%, Сmax 16%). Степень взаимодействия не считается клинически значимой, и, таким образом, коррекции дозы субстратов CYP3A не требуется. Согласно фармакокинетическому моделированию, основанному на физиологии, ожидается, что данный эффект будет иметь сходную выраженность у младенцев старше 2 месяцев и детей.
Исследования in vitro показали, что рисдиплам и его основной метаболит не являются значимыми ингибиторами MDR1, транспортного полипептида органических анионов ОАТР1В1 и ОАТР1В3 и переносчика органических анионов ОАТ1 и ОАТ3 у человека.
Рисдиплам и его метаболит, однако, являются ингибиторами переносчика органических катионов ОСТ2, белка экструзии лекарственных препаратов и токсинов МАТЕ1 и МАТЕ2-К в условиях in vitro. При терапевтических концентрациях препарата взаимодействий с субстратами ОСТ2 не ожидается. Согласно данным, полученным в условиях in vitro, при применении препарата Эврисди® плазменные концентрации препаратов, которые выводятся посредством МАТЕ1 и МАТЕ2-К, могут повышаться. Клиническая значимость одновременного применения с субстратами МАТЕ1/2-К неизвестна.
Особые указания
Эмбриофетальная токсичность
При изучении применения препарата у животных наблюдалась эмбриофетальная токсичность. Пациенты репродуктивного потенциала должны быть проинформированы о рисках и использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приема последней дозы препарата Эврисди® пациентом-женщиной и в течение, как минимум, 4 месяцев после приема последней дозы препарата Эврисди® пациентом-мужчиной (см. раздел «Применение при беременности и в период грудного вскармливания»).
Возможное влияние на фертильность у мужчин
Обратимое влияние препарата Эврисди® на мужскую фертильность было показано в исследовании у животных. В связи с этим, пациенты-мужчины не должны быть донорами спермы во время лечения и в течение 4 месяцев после приема последней дозы препарата Эврисди® (см. раздел «Применение при беременности и в период грудного вскармливания»).
Нарушение функции печени
Фармакокинетику, безопасность и переносимость рисдиплама в дозе 5 мг оценивали у субъектов с нарушением функции печени легкой или средней степени тяжести в специальном клиническом исследовании.
Нарушение функции печени легкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. Таким образом, пациентам с нарушением функции печени легкой или средней степени тяжести коррекции дозы не требуется. Применение препарата Эврисди® у пациентов с нарушением функции печени тяжелой степени тяжести не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов» и раздел «Способ применения и дозы», подраздел «Дозирование в особых случаях»).
Злоупотребление приемом препарата или лекарственная зависимость
Препарат Эврисди® не обладает потенциалом, который может привести к злоупотреблению приемом препарата и лекарственной зависимости.
Влияние на способность управлять транспортными средствами, механизмами
Препарат Эврисди® не оказывает влияния на способность управлять транспортными средствами, механизмами.
Форма выпуска
Порошок для приготовления раствора для приема внутрь 0.75 мг/мл
По 2 г порошка во флакон темного стекла (гидролитический класс III Ф.США/ЕФ) с завинчивающейся пластиковой крышкой, оснащенной уплотнительным диском, с защитой от детей и кольцом первого вскрытия.
1 флакон вместе с размещенными в отдельном прозрачном пакете адаптером из полиэтилена, 2 пероральными шприцами из полипропилена вместимостью 6 мл (помещенными в непрозрачный пакет с маркировкой, обозначающей объем дозирования),
2 пероральными шприцами из полипропилена вместимостью 12 мл (помещенными в непрозрачный пакет с маркировкой, обозначающей объем дозирования), инструкцию по применению и руководство по разведению (только для медицинских работников) помещают в картонную пачку.
С целью контроля первого вскрытия на пачку наносится защитная голографическая наклейка.
Срок годности
2 года.
Не применять по истечении срока годности.
Приготовленный раствор хранить в оригинальном флаконе с плотно закрытой крышкой в вертикальном положении при температуре 2-8 °С в течение 64 дней, не замораживать.
Условия хранения
При температуре не выше 25 °С в оригинальной упаковке (флакон в картонной пачке).
Условия отпуска
Отпускают по рецепту.
Владелец регистрационного удостоверения
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Производитель
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Фасовщик (первичная упаковка)
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, 4070 Basel, Switzerland
Упаковщик (вторичная/потребительская упаковка)
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg, 4303 Kaiseraugst, Switzerland
Выпускающий контроль качества
Ф. Хоффманн-Ля Рош Лтд., Швейцария
F. Hoffmann-La Roche Ltd, Wurmisweg, 4303 Kaiseraugst, Switzerland
Организация, принимающая претензии от потребителей
Претензии потребителей направлять в компанию АО «Рош-Москва» по адресу:
107045, Россия, г. Москва, Трубная площадь, д. 2, помещение 1, этаж 1, комната 42
Руководство по разведению
препарата ЭВРИСДИ® (рисдиплам) в виде раствора для приема внутрь 0.75 мг/мл
Только для медицинских работников
Каждая картонная пачка препарата Эврисди® содержит (см. рисунок А):
- Крышка флакона – 1 шт.
- Флакон с препаратом Эврисди® – 1 шт.
- Пероральные шприцы на 12 мл (в пакетах) – 2 шт.
- Пероральные шприцы на 6 мл (в пакетах) – 2 шт.
- Адаптер для флакона (вдавливаемый) – 1 шт.
- Инструкция по медицинскому применению (не показана на рисунке) – 1 шт.
- Руководство по разведению (не показано на рисунке) – 1 шт.
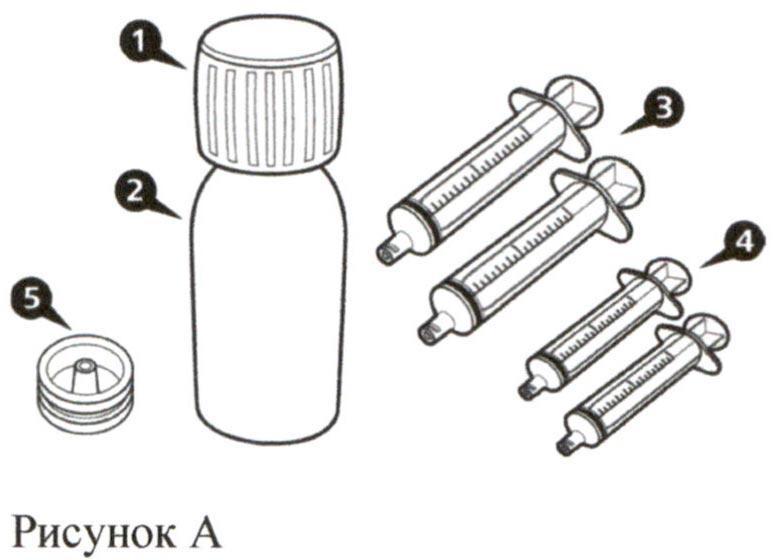
Важная информация по препарату Эврисди®
- Избегайте вдыхания порошка препарата Эврисди®.
- Используйте перчатки.
- Не используйте порошок, если его срок годности истек. Дата истечения срока годности (Годен до) порошка напечатана на этикетке флакона.
- Не выдавайте приготовленный раствор, если дата утилизации раствора наступит позже, чем дата истечения срока годности исходного порошка.
- Избегайте контакта препарата с Вашей кожей. Если это произошло, промойте данную область водой с мылом.
- Не используйте препарат, если какие-либо элементы упаковки повреждены или отсутствуют.
- Используйте воду очищенную или стерильную воду для инъекций для разведения препарата.
- Не добавляйте другие пероральные шприцы, помимо предоставляемых в картонной пачке.
Как хранить препарат Эврисди®
- Порошок (неразведенный препарат) хранить при комнатной температуре не выше 25 °С в картонной пачке.
- Приготовленный раствор (разведенный препарат) хранить в холодильнике при температуре 2-8 °С.
- Приготовленный раствор для приема внутрь хранить в оригинальном флаконе с плотно закрытой крышкой в вертикальном положении.
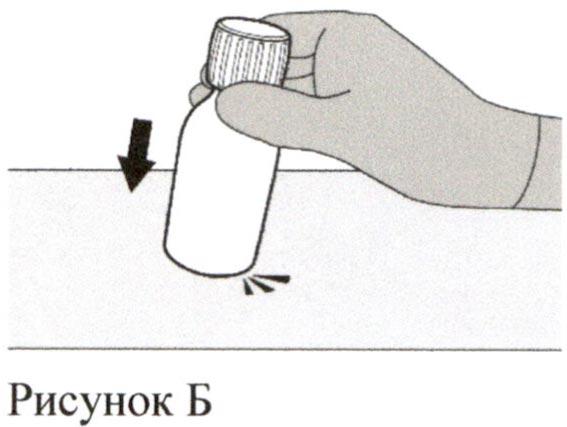
Разведение Шаг 1
Аккуратно постучите по дну флакона для разрыхления порошка (см. Рисунок Б).
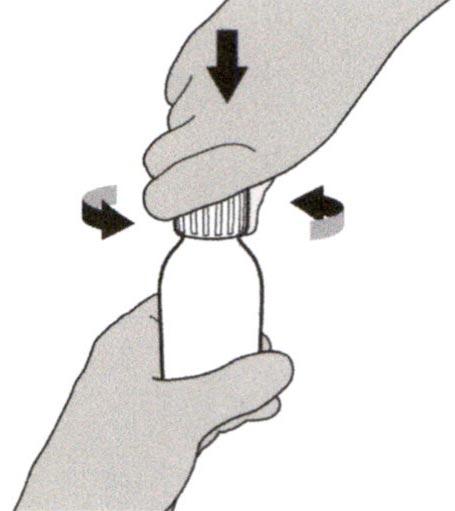
Шаг 2
Снимите крышку, нажав на нее вниз и затем повернув налево (против часовой стрелки) (см. Рисунок В). Не выбрасывайте крышку.
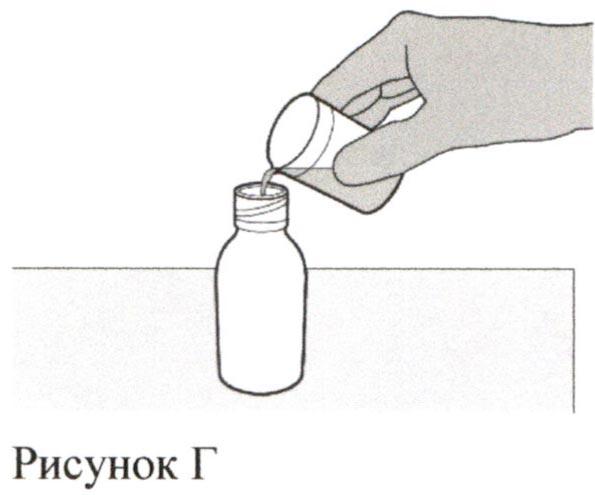
Шаг 3
Аккуратно налейте 79 мл воды очищенной или стерильной воды для инъекций во флакон с препаратом (см. Рисунок Г).
Шаг 4
Удерживайте флакон с препаратом на столе одной рукой.
Вставьте вдавливаемый адаптер для флакона в горлышко флакона, надавливая на адаптер вниз другой рукой. Убедитесь, что адаптер для флакона полностью вдавлен в горлышко флакона (см. Рисунок Д).
Шаг 5
Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы закрыть флакон.
Убедитесь, что флакон полностью закрыт, и затем хорошо встряхивайте его в течение 15 секунд (см. Рисунок Е).
Подождите 10 минут. У Вас должен получиться прозрачный раствор.
Если этого не произошло, следует хорошо встряхивать флакон в течение 15 секунд еще раз.
Шаг 6
Рассчитайте дату утилизации приготовленного раствора для приема внутрь, равную 64 дням после разведения (Примечание: день разведения считается днем 0. Например, если разведение произведено 1 апреля, датой утилизации будет 4 июня).
Запишите дату утилизации приготовленного раствора на этикетке флакона (см. рисунок Ж) и на картонной пачке.
Поместите флакон обратно в оригинальную картонную пачку вместе со шприцами (в пакетах) и инструкцией по медицинскому применению. Хранить картонную пачку в холодильнике.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)
Risdiplamum (род. Risdiplami)
7-(4,7-диазаспиро[2.5]октан-7-ил)-2-(2,8-диметилимидазо[1,2-b]пиридазин-6-ил)пиридо[1,2-a]пиримидин-4-он
Рисдиплам — модификатор сплайсинга РНК гена выживаемости двигательных нейронов 2 (SMN2).
Молекулярная масса 401,46 г/моль.
Механизм действия
Рисдиплам — модификатор сплайсинга РНК гена выживаемости двигательных нейронов 2 (SMN2), разработанный для лечения пациентов со спинальной мышечной атрофией (СМА), вызванной мутациями в хромосоме 5q, которые приводят к дефициту белка SMN. Используя тесты in vitro и исследования на трансгенных животных моделях СМА, было показано, что рисдиплам увеличивает включение экзона 7 в транскрипты рибонуклеиновой кислоты (мРНК) SMN2 и образование полноразмерного белка SMN в мозге.
Данные in vitro и in vivo показывают, что рисдиплам может вызывать альтернативный сплайсинг дополнительных генов, включая FOXM1 и MADD. Считается, что FOXM1 и MADD участвуют в регуляции клеточного цикла и апоптоза соответственно и были идентифицированы как возможные участники развития неблагоприятных эффектов, наблюдаемых у животных.
Фармакодинамика
В клинических испытаниях рисдиплам приводил к увеличению уровня белка SMN, и в течение 4 нед после начала лечения медианный уровень белка SMN был более чем в 2 раза выше по сравнению с исходным. Увеличение продолжалось на протяжении всего периода лечения (не менее 12 мес) при всех типах СМА.
Фармакокинетика
Фармакокинетика рисдиплама была охарактеризована у здоровых взрослых людей и пациентов со СМА.
После приема в виде перорального раствора фармакокинетика рисдиплама была приблизительно линейной в диапазоне от 0,6 до 18 мг в исследовании с однократной возрастающей дозой у здоровых взрослых людей и от 0,02 до 0,25 мг/кг при приеме 1 раз в день в многократной возрастающей дозе в исследовании у пациентов со СМА. После перорального приема рисдиплама 1 раз в день у здоровых людей наблюдалось примерно 3-кратное увеличение Cmax в плазме и AUC0–24. Экспозиция рисдиплама достигала равновесного состояния через 7–14 дней после приема 1 раз в сутки.
Абсорбция
После приема внутрь Tmax в плазме составляет от 1 до 4 ч.
Распределение
Кажущийся Vss составляет 6,3 л/кг.
Рисдиплам преимущественно связывается с сывороточным альбумином, без какого-либо связывания с кислым альфа1-гликопротеином, свободная фракция составляет 11%.
Элиминация
Кажущийся клиренс (CL/F) рисдиплама — 2,1 л/ч для пациента с массой тела 14,9 кг.
Конечный T1/2 рисдиплама у здоровых взрослых — приблизительно 50 ч.
Метаболизм. Рисдиплам в основном метаболизируется флавинмонооксигеназой 1 и 3 (FMO1 и FMO3), а также изоферментами CYP1A1, CYP2J2, CYP3A4 и CYP3A7.
Исходный рисдиплам был основным компонентом, обнаруженным в плазме, на его долю приходилось 83% от общих компонентов ЛС, находящихся в кровотоке. Фармакологически неактивный метаболит M1 был идентифицирован как основной циркулирующий метаболит.
Выведение. После приема 18 мг примерно 53% дозы (14% неизмененного рисдиплама) выводится с фекалиями и 28% — с мочой (8% неизмененного рисдиплама).
Особые группы пациентов
Не было клинически значимых различий фармакокинетики рисдиплама в зависимости от расы или пола. Не ожидается, что нарушение функции почек будет оказывать влияние на экспозицию рисдиплама.
Влияние гериатрического возраста на фармакокинетику рисдиплама не изучали.
Нарушение функции печени. Фармакокинетика и безопасность рисдиплама были изучены у людей с легкой или умеренной печеночной недостаточностью (по классификации Чайлд-Пью класса A и B соответственно, n=8 для каждого) по сравнению с людьми с нормальной функцией печени (n=10). После приема рисдиплама в дозе 5 мг AUCinf и Cmax рисдиплама изменялись примерно на 20 и 5% соответственно у пациентов с легкой печеночной недостаточностью и на 8 и 20% соответственно у пациентов с умеренной печеночной недостаточностью по сравнению с подобранными здоровыми добровольцами контрольной группы. Величина этих изменений не считается клинически значимой. Фармакокинетика и безопасность у пациентов с тяжелой печеночной недостаточностью (класс C по шкале Чайлд-Пью) не изучались.
Педиатрические пациенты. Было обнаружено, что масса тела и возраст оказывают значительное влияние на фармакокинетику рисдиплама. Расчетная экспозиция (значение AUC0–24) для пациентов с манифестацией СМА в младенческом возрасте (от 2 до 7 мес на момент включения в исследование) при рекомендуемой дозе 0,2 мг/кг 1 раз в день составляла 1930 нг·ч/мл. Расчетная экспозиция для пациентов с поздней манифестацией СМА (от 2 до 25 лет на момент включения в исследование) при рекомендованной дозе составляла 2050 нг·ч/мл (0,25 мг/кг 1 раз в день для пациентов с массой тела <20 кг и 5 мг 1 раз в день для пациентов с массой тела ≥20 кг). Наблюдаемая средняя Cmax составляла 184 нг/мл для пациентов c манифестацией СМА в младенческом возрасте и 148 нг/мл для пациентов с поздней манифестацией СМА.
Согласно литературным данным, ожидается, что у педиатрических пациентов в возрасте до 2 мес снижена активность FMO3, что может привести к увеличению экспозиции рисдиплама (см. Элиминация).
Нет данных о фармакокинетике рисдиплама у пациентов в возрасте до 2 мес (см. «Меры предосторожности»).
Доклиническая токсикология
Канцерогенность, мутагенность, влияние на фертильность
Канцерогенность. Канцерогенный потенциал рисдиплама полностью не изучен. Рисдиплам не оказывал канцерогенного действия у мышей линии Tg.rasH2 при пероральном применении в дозах до 9 мг/кг/сут в течение 26 нед.
Мутагенность. In vitro тест Эймса для рисдиплама был негативным. В комбинированном in vivo микроядерном тесте на клетках костного мозга и ДНК-комет у крыс рисдиплам проявлял кластогенность, о чем свидетельствует увеличение количества микроядер в костном мозге, но оказался негативным в анализе ДНК-комет. Выраженное увеличение микроядер костного мозга также наблюдалось в исследованиях токсичности у взрослых и молодых крыс (см. «Меры предосторожности»).
Влияние на фертильность. Пероральное применение рисдиплама у крыс в течение 4 (0; 1; 3 или 9 мг/кг/сут) или 26 (0; 1; 3 или 7,5 мг/кг/сут) нед приводило к гистопатологическим изменениям в яичках (дегенерация сперматоцитов, дегенерация/атрофия семенных канальцев) и придатках яичек (дегенерация/некроз эпителия протоков) при средних и/или высоких дозах. При высокой дозе в 26-недельном исследовании повреждения яичек сохранялись до конца периода восстановления, что соответствует у крыс примерно одному сперматогенному циклу. Доза, не вызывающая неблагоприятных воздействий на репродуктивную систему, у взрослых крыс-самцов (1 мг/кг/сут) была ассоциирована с экспозицией ЛС в плазме (AUC), сходной с таковой у человека при МРДЧ 5 мг/сут.
Неблагоприятные эффекты рисдиплама на семенники не могли быть полностью оценены у обезьян, потому что большинство протестированных обезьян были неполовозрелыми. Однако пероральное применение рисдиплама (0; 2; 4 или 6 мг/кг/сут) в течение 2 нед приводило к гистопатологическим изменениям в яичках (увеличение количества многоядерных клеток, дегенерация половых клеток) при самой высокой дозе. При дозе, не вызывающей тестикулярную токсичность у обезьян, плазменная экспозиция была примерно в 3 раза выше, чем у человека при МРДЧ.
Пероральное применение рисдиплама у молодых крыс после прекращения вскармливания приводило к токсичности для репродуктивной системы самцов (дегенерация/некроз сперматогенного эпителия яичек с ассоциированной олиго/аспермией в придатке яичка и аномальные параметры спермы). Доза, не вызыващая неблагоприятных воздействий на репродуктивную систему, у молодых крыс-самцов после прекращения вскармливания была ассоциирована с плазменной экспозицией примерно в 4 раза выше, чем у человека при МРДЧ (см. «Меры предосторожности»).
Фармакология и/или токсикология у животных
Ретинальная токсичность
В исследованиях на животных были обнаружены индуцированные рисдипламом функциональные и структурные аномалии сетчатки. В 39-недельном исследовании токсичности у обезьян пероральный прием рисдиплама (0; 1,5; 3 или 7,5/5 мг/кг/сут; высокая доза была снижена через 4 нед) вызывал функциональные нарушения на электроретинограмме (ЭРГ) при всех средних и высоких дозах у животных в самое раннее время обследования (20-я нед). Эти нарушения были ассоциированы с дегенерацией сетчатки, обнаруженной с помощью оптической когерентной томографии (ОКТ) на 22-й нед, при первом обследовании. Дегенерация сетчатки с потерей периферических фоторецепторов была необратимой. Доза, не вызывающая воздействий на изменения в сетчатке (1,5 мг/кг/сут) была ассоциирована с плазменной экспозицией (AUC), аналогичной таковой у человека при МРДЧ 5 мг.
Влияние на эпителиальные ткани
Пероральное применение рисдиплама у крыс и обезьян приводило к гистопатологическим изменениям эпителия ЖКТ (апоптоз/некроз единичных клеток), собственной пластинки (вакуолизация), экзокринной части поджелудочной железы (некроз единичных клеток), кожи, языка и гортани (паракератоз/гиперплазия/дегенерация) с сопутствующим воспалением. Влияние на эпителий кожи и ЖКТ было обратимым. Дозы, не вызывающие воздействий на эпителиальные ткани у крыс и обезьян, были ассоциированы с плазменной экспозицией (AUC), аналогичной таковой у человека при МРДЧ.
Клинические исследования
Эффективность рисдиплама для лечения пациентов с манифестацией СМА в младенческом возрасте и пациентов с поздней манифестацией СМА оценивалась в двух клинических исследованиях, исследовании 1 (NCT02913482) и исследовании 2 (NCT02908685).
В целом результаты этих исследований подтверждают эффективность рисдиплама у пациентов со СМА в возрасте 2 мес и старше и являются основанием для раннего начала лечения рисдипламом.
Манифестация СМА в младенческом возрасте
Исследование 1 — открытое, состоящее из двух частей исследование, посвященное изучению эффективности, безопасности, фармакокинетики и фармакодинамики рисдиплама у пациентов со СМА I типа (появление симптомов в возрасте от 28 дней до 3 мес). В части 1 исследования 1 (n=21) представлены данные об эффективности и безопасности у пациентов со СМА I типа. Дополнительная информация о безопасности предоставлена в части 2 исследования 1 (n=41) у пациентов со СМА I типа (см. «Побочные действия»).
В части 1 исследования 1 пациенты (n=21) были включены в одну из двух когорт дозирования. Пациентам в когорте с более высокими дозами (n=17) доза была скорректирована до рекомендуемой 0,2 мг/кг/сут до 12 мес лечения, в то время как у пациентов в группе с низкой дозой (n=4) этого не делали.
Эффективность была установлена на основе возможности сидеть без поддержки в течение как минимум 5 с (согласно измерениям по пункту 22 Bayley Scales of Infant and Toddler Development — Third Edition (BSID-III) gross motor scale) (шкала развития младенцев и детей Бейли — 3-е издание, шкала крупной моторики) и на основе выживаемости без необходимости постоянной вентиляции. Постоянная вентиляция была определена как требующая трахеостомии или более 21 дня подряд, либо неинвазивной вентиляции (≥16 ч в день), либо интубации при отсутствии острого обратимого события.
Медиана возраста возникновения клинических признаков и симптомов СМА I типа у пациентов, включенных в часть 1 исследования 1, составила 2 мес (диапазон от 0,9 до 3), 71% пациентов были женского пола, 81% — представители европеоидной расы и 19% — представители азиатской расы. Медиана возраста на момент набора в исследование составила 6,7 мес (диапазон от 3,3 до 6,9), и медиана времени между появлением симптомов и приемом первой дозы составила 4 мес (диапазон от 2 до 5,8). У всех пациентов было генетически подтверждено наличие гомозиготной делеции или сложной гетерозиготности, прогнозирующих потерю функции гена SMN1, и две копии гена SMN2.
В исследовании 1, часть 1, медиана продолжительности лечения рисдипламом составила 14,8 мес (диапазон от 0,6 до 26), и у 19 пациентов минимальная продолжительность лечения составила 12 мес.
Из пациентов, получавших рекомендованную дозу рисдиплама 0,2 мг/кг/сут, 41% (7/17) смогли самостоятельно сидеть в течение ≥5 с (BSID-III, пункт 22) после 12 мес лечения. Эти результаты указывают на клинически значимое отклонение от естественного течения нелеченой СМА с манифестацией в младенческом возрасте. Как описано для естественного течения нелеченой СМА с манифестацией в младенческом возрасте, не ожидается, что пациенты смогут самостоятельно сидеть, и не более 25% этих пациентов выживут без постоянной вентиляции после достижения 14-месячного возраста. После 12 мес лечения рисдипламом 90% (19/21) пациентов остались живы без необходимости постоянной вентиляции (и достигли возраста 15 мес и старше). После как минимум 23 мес лечения рисдипламом 81% (17/21) все пациенты были живы и не нуждались в постоянной вентиляции (и достигли возраста 28 мес и старше; в среднем 32 мес; диапазон от 28 до 45 мес).
Поздняя манифестация СМА
Исследование 2 — многоцентровое исследование, состоящее из двух частей, по изучению эффективности, безопасности, фармакокинетики и фармакодинамики рисдиплама у пациентов с диагнозом СМА II или III типа. Часть 1 исследования 2 была разработана как часть исследования по подбору дозы у 51 пациента (14% амбулаторно). Часть 2 была рандомизированной двойной слепой плацебо-контролируемой и описана ниже.
Первичной конечной точкой исследования 2, часть 2, было изменение показателя Motor Function Measure 32 (MFM32) score, шкала измерения моторных функций 32, MFM32), от исходного уровня до 12 мес. Ключевой вторичной конечной точкой была доля пациентов с изменением на 3 или более балла от исходного уровня до 12 мес по общему баллу MFM32. MFM32 позволяет оценить двигательные функции, относящиеся к повседневным функциям. Общий балл MFM32 выражается в процентах (диапазон от 0 до 100) от максимально возможного балла, причем более высокие баллы указывают на бóльшую двигательную функцию. Другой ключевой вторичной конечной точкой была оценка по Revised Upper Limb Module, обновленный модуль оценки моторной функции верхних конечностей, RULM). RULM — это инструмент, используемый для оценки двигательной активности верхних конечностей у пациентов со СМА. Шкала дает оценку проксимальной и дистальной двигательных функций руки. Общий балл варьирует от 0 (все пункты не могут быть выполнены) до 37 (все действия выполняются полностью без каких-либо компенсирующих ходов).
В исследование 2, часть 2, были включены 180 неамбулаторных пациентов со СМА II типа (71%) или III типа (29%). Пациенты были рандомизированы 2:1 для получения рисдиплама в рекомендованной дозе или плацебо. Рандомизация была стратифицирована по возрастным группам (от 2 до 5, от 6 до 11, от 12 до 17 или от 18 до 25 лет).
Медиана возраста пациентов на момент начала лечения составила 9 лет (от 2 до 25), а медиана времени между появлением первых симптомов СМА и приемом первой дозы составила 102,6 мес (от 1 до 275). Из 180 пациентов, включенных в исследование, 51% были женского пола, 67% — представители европеоидной расы и 19% — представители азиатской расы. Исходно у 67% пациентов был выявлен сколиоз (у 32% из них — тяжелый сколиоз). Среднее значение MFM32 у пациентов на исходном уровне составляло 46,1, а по RULM — 20,1. В целом демографические характеристики пациентов на исходном уровне были хорошо сбалансированы между группами (рисдиплам и плацебо), за исключением сколиоза (63% в группе, получавшей рисдиплам, по сравнению с 73% в группе, получавшей плацебо).
Первичный анализ изменения общего балла MFM32 по сравнению с исходным уровнем на 12-м мес показал клинически и статистически значимое различие между пациентами, получавшими рисдиплам и получавшими плацебо. Результаты первичного анализа и ключевые вторичные конечные точки показаны в таблице 1.
Таблица 1
Сводная информация данных по эффективности у пациентов с поздней манифестацией СМА на 12-м мес лечения (исследование 2, часть 2)
| Конечная точка | Рисдиплам (n=120) | Плацебо (n=60) |
| Первичная конечная точка | ||
| Изменение общего балла MFM32 по сравнению с исходным уровнем на 12-м мес, LS значение (95% ДИ)1,2,3 | 1,36 (0,61; 2,11) | −0,19 (−1,22; 0,84) |
| Отличие от плацебо, расчетное значение (95% ДИ)1, p-значение | 1,55 (0,3; 2,81), 0,0156 | |
| Вторичные точки | ||
| Доля пациентов с изменением общего балла MFM32 по сравнению с исходным уровнем на 3 или более на 12-м мес (95% ДИ)2,3 | 38,3% (28,9; 47,6) | 23,7% (12; 35,4) |
| Отношение шансов для общего ответа (95% ДИ), скорректированное4 p-значение5 (без коррекции) | 2,35 (1,01; 5,44), 0,469 (0,0469) | |
| Изменение общего балла RULMat на 12-м мес от исходного уровня, LS значение (95% ДИ)1,6 | 1,61 (1; 2,22) | 0,02 (−0,83; 0,87) |
| Отличие от плацебо, расчетное значение (95% ДИ)1, скорректированное4 p-значение1 (без коррекции) | 1,59 (0,55; 2,62), 0,0469 (0,0028) |
LS значение — среднее значение, рассчитанное по методу наименьших квадратов.
1 Анализ Mixed Model Repeated Measure (смешанная модель повторных измерений, MMRM) включал изменение от исходного уровня общего балла как зависимую переменную, и как независимые переменные — общий балл на исходном уровне, группу лечения, время, зависимость эффекта лечения от времени и переменную стратификации рандомизации групп по возрасту (от 2 до 5, от 6 до 11, от 12 до 17, от 18 до 25).
2 Общий балл MFM был рассчитан в соответствии с руководством пользователя и выражен как процент от максимально возможного балла по шкале (т.е. сумма баллов по 32 пунктам, разделенная на 96 и умноженная на 100).
3 На основании правила отсутствия данных для MFM32 из анализа были исключены шесть пациентов (рисдиплам, n=115; плацебо-контроль, n=59).
4 Скорректированное значение p было получено для конечных точек, включенных в иерархическое тестирование, и было основано на всех значениях p от конечных точек в порядке иерархии до текущей конечной точки.
5 Анализ логистической регрессии включал исходный общий балл, лечение и возрастную группу в качестве независимых переменных.
6 На основании правила отсутствия данных для RULM, три пациента были исключены из анализа (рисдиплам, n=119; плацебо-контроль, n=58).
Лечение спинальной мышечной атрофии у пациентов в возрасте 2 мес и старше.
Нет.
Беременность
Резюме рисков
Нет адекватных данных о риске развития, связанном с применением рисдиплама у беременных женщин. В исследованиях на животных применение рисдиплама во время беременности или на протяжении беременности и лактации приводило к неблагоприятным последствиям для развития (эмбриофетальная смертность, пороки развития, снижение массы тела плода и нарушение репродуктивной функции у потомства) при клинически значимой или выше экспозиции ЛС (см. Данные).
Предполагаемый фоновый риск серьезных врожденных дефектов и выкидыша для указанной популяции неизвестен. Для населения США в целом оценочный фоновый риск серьезных врожденных дефектов и выкидыша при клинически признанных беременностях составляет от 2 до 4% и от 15 до 20% соответственно. Основываясь на данных у животных, необходимо сообщить беременной женщине о потенциальном риске для плода.
Данные
Данные у животных. Пероральное применение рисдиплама (0; 1; 3 или 7,5 мг/кг/сут) у беременных крыс на протяжении периода органогенеза приводило к снижению массы тела плодов и увеличению частоты структурных изменений плодов при наивысшей тестируемой дозе, что не было ассоциировано с материнской токсичностью. Уровень, не вызывающий неблагоприятных эффектов на эмбриофетальное развитие (3 мг/кг/сут), был ассоциирован с экспозицией в материнской плазме (AUC) примерно в 2 раза выше, чем у человека при МРДЧ 5 мг.
Пероральное применение рисдиплама (0; 1; 4 или 12 мг/кг/сут) у беременных крольчих на протяжении органогенеза приводило к эмбриофетальной смертности, порокам развития плода (гидроцефалия) и структурным изменениям при самой высокой испытанной дозе, что было ассоциировано с материнской токсичностью. Доза, не вызывающая неблагоприятных эффектов на эмбриофетальное развитие (4 мг/кг/сут) была ассоциирована с с экспозицией в материнской плазме (AUC) примерно в 4 раза выше, чем у человека при МРДЧ.
При пероральном применении рисдиплама (0; 0,75; 1,5 или 3 мг/кг/сут) у крыс на протяжении беременности и лактации беременность у самок продлевалась, замедлялось половое созревание (открытие влагалища) и нарушалась репродуктивная функция (снижение числа желтых тел, мест имплантации и числа живых эмбрионов) у потомства крыс при максимальной дозе. Доза, не вызывающая неблагоприятных эффектов на пре- и постнатальное развитие у крыс (1,5 мг/кг/сут) была ассоциирована с экспозицией материнской плазмы (AUC), сходной с таковой у человека при МРДЧ.
Грудное вскармливание
Резюме рисков
Нет данных о присутствии рисдиплама в женском молоке, его влиянии на грудного ребенка или влиянии на выработку молока. Рисдиплам экскретировался в молоко лактирующих крыс, которые получали рисдиплам перорально.
Следует учитывать преимущества грудного вскармливания для развития и здоровья, а также клиническую потребность матери в рисдипламе и любые потенциальные неблагоприятные эффекты рисдиплама для грудного ребенка или основное состояние матери.
Репродуктивный потенциал мужчин и женщин
Исследования рисдиплама у молодых и взрослых крыс и обезьян продемонстрировали неблагоприятное воздействие на репродуктивные органы, включая половые клетки мужчин, при клинически значимых плазменных экспозициях (см. «Меры предосторожности» и Доклиническая токсикология).
Тест на беременность
Перед началом приема рисдиплама женщинам с репродуктивным потенциалом рекомендуется пройти тестирование на беременность.
Контрацепция
Рисдиплам может вызывать повреждение эмбриона и плода при применении беременной женщиной.
Пациенты женского пола. Необходимо посоветовать пациенткам с репродуктивным потенциалом использовать эффективную контрацепцию во время лечения рисдипламом и в течение как минимум 1 мес после последней дозы.
Бесплодие
Пациенты мужского пола. Мужская фертильность может быть снижена при лечении рисдипламом (см. Доклиническая токсикология).
Опыт клинических испытаний
Поскольку клинические испытания проводятся с различным набором условий, частота встречаемости побочных реакций, наблюдаемых в этих испытаниях, не может непосредственно сравниваться с частотой в других клинических испытаниях и может не отражать наблюдаемую в клинической практике.
В клинических испытаниях с участием пациентов с манифестацией СМА в младенческом возрасте и поздней манифестацией СМА в общей сложности 337 пациентов (52% женского пола, 72% европеоидной расы) принимали рисдиплам сроком максимум 32 мес, при этом 209 пациентов получали лечение в течение более 12 мес. Сорок семь (14%) пациентов были в возрасте 18 лет и старше, 74 (22%) — от 12 до 18 лет, 154 (46%) — от 2 до 12 лет и 62 (18%) — от 2 мес до менее 2 лет.
Клинические испытания при поздней манифестации СМА
Безопасность рисдиплама при СМА с поздним началом основана на данных рандомизированного двойного слепого плацебо-контролируемого исследования (исследование 2, часть 2) у пациентов со СМА II или III типа (n=180) (см. Клинические исследования). Возраст в популяции пациентов в исследовании 2, часть 2, составлял в от 2 до 25 лет на момент начала лечения.
Наиболее частыми побочными реакциями (зарегистрированными не менее чем у 10% пациентов, получавших рисдиплам, и с частотой выше, чем в группе плацебо) в исследовании 2, часть 2, были лихорадка, диарея и сыпь. В таблице 2 перечислены побочные реакции, которые наблюдались по крайней мере у 5% пациентов, получавших рисдиплам, и с частотой ≥5%, чем в группе плацебо, в исследовании 2, часть 2.
Таблица 2
Побочные реакции, зарегистрированные у ≥5% пациентов, получавших рисдиплам, с частотой ≥5%, чем при применении плацебо, в исследовании 2, часть 2
| Побочная реакция | Рисдиплам (n=120), % | Плацебо (n=60), % |
| Лихорадка1 | 22 | 17 |
| Диарея | 17 | 8 |
| Сыпь2 | 17 | 2 |
| Язвы во рту и афтозные язвы | 7 | 0 |
| Артралгия | 5 | 0 |
| Инфекция мочевыводящих путей3 | 5 | 0 |
1 Включает пирексию и гиперпирексию.
2 Включает сыпь, эритему, макулопапулезную сыпь, эритематозную сыпь, папулезную сыпь, аллергический дерматит и фолликулит.
3 Включает инфекцию мочевыводящих путей и цистит.
Клинические испытания при манифестации СМА в младенческом возрасте
Безопасность терапии рисдиплама при манифестации СМА в младенческом возрасте основана на данных открытого исследования с участием 62 пациентов (исследование 1) (см. Клинические исследования). В исследовании 1, часть 1 (n=21) и часть 2 (n=41), 62 пациента получали рисдиплам сроком до 30 мес (31 пациент более 12 мес). Возраст пациентов на момент начала лечения составлял от 2 до 7 мес (масса тела от 4,1 до 10,6 кг).
Наиболее частые побочные реакции, отмеченные у пациентов при манифестации СМА в младенческом возрасте, получавших рисдиплам в исследовании 1, были сходны с теми, которые наблюдались у пациентов со СМА с поздним началом в исследовании 2. Кроме того, следующие побочные реакции были зарегистрированы у ≥10% пациентов: инфекция верхних дыхательных путей (включая назофарингит, ринит, инфекцию дыхательных путей), пневмония, запор и рвота.
Влияние других ЛС на рисдиплам
Совместное применение 200 мг итраконазола (сильный ингибитор CYP3A) 2 раза в день с однократной пероральной дозой 6 мг рисдиплама не оказывало клинически значимого влияния на фармакокинетику рисдиплама (увеличение AUC на 11% и снижение Cmax на 9%).
Рисдиплам является слабым субстратом белка множественной лекарственной устойчивости (multidrug resistance, MDR1) у человека и переносчика BCRP in vitro. Ожидается, что ингибиторы MDR1 или BCRP не приведут к клинически значимому увеличению концентрации рисдиплама.
Влияние рисдиплама на другие ЛС
Рисдиплам и его основной циркулирующий метаболит M1 не индуцировали изоферменты CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 или CYP3A4 in vitro. Рисдиплам и M1 не ингибировали (обратимое или зависящее от времени ингибирование) ни один из тестируемых изоферментов CYP (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6), за исключением CYP3A in vitro.
Рисдиплам — слабый ингибитор CYP3A. У здоровых взрослых людей применение рисдиплама 1 раз в день в течение 2 нед несколько увеличивало экспозицию мидазолама, чувствительного субстрата CYP3A (AUC 11%, Cmax 16%), это увеличение не считается клинически значимым. На основе физиологически обоснованного фармакокинетического моделирования аналогичное увеличение ожидается как у маленьких детей, так и у младенцев в возрасте 2 мес.
Исследования in vitro показали, что рисдиплам и его основной метаболит не являются значительными ингибиторами MDR1 человека, транспортеров OATP1B1, OATP1B3, OAT1 и -3 и OCT2 в клинически значимых концентрациях. Однако рисдиплам и его метаболит in vitro являются ингибиторами транспортеров MATE1 и MATE2-K.
Влияние рисдиплама на субстраты транспортеров MATE. Согласно данным in vitro, рисдиплам может увеличивать плазменные концентрации ЛС, элиминирующихся посредством MATE1 или MATE2-K, таких как метформин. Следует избегать сочетанного применения рисдиплама с субстратами MATE. При невозможности избежать совместного применения необходим мониторинг токсичности, связанной с ЛС, и при необходимости следует рассмотреть возможность снижения дозы совместно принимаемого ЛС (на основе инструкции этого ЛС).
Информация не предоставлена.
Внутрь, 1 раз в сутки, доза зависит от возраста и массы тела.
Применение в педиатрии
Безопасность и эффективность рисдиплама у педиатрических пациентов в возрасте 2 мес и старше были установлены (см. Клинические исследования). Безопасность и эффективность у педиатрических пациентов в возрасте до 2 мес не были установлены (см. «Фармакология»).
Данные о токсичности у неполовозрелых животных. Пероральное применение рисдиплама (0; 0,75; 1,5; 2,5 мг/кг/сут) у молодых крыс с 4-го по 31-й постнатальный день (ПНД) приводило к снижению роста (масса тела, длина голени) и задержке полового созревания у самцов при средней и высокой дозах. Недостаточность развития скелета и массы тела сохранялись после прекращения дозирования. При высокой дозе наблюдались офтальмологические изменения, состоящие из вакуолей в передней части стекловидного тела. Снижение абсолютного количества В-лимфоцитов наблюдалось при всех дозах после прекращения дозирования. Уменьшение веса яичек и придатков яичка, которое коррелировало с дегенерацией семенного эпителия в яичках, происходило при средних и высоких дозах; результаты гистопатологии были обратимыми, но вес органа сохранялся после прекращения дозирования. Нарушения репродуктивной функции у самок (снижение индекса спаривания, индекса фертильности и частоты зачатия) наблюдались при высокой дозе. Доза, не оказывающая влияния на возникновение побочных эффектов развития у крыс до прекращения вскармливания, не была идентифицирована. Самая низкая испытанная доза (0,75 мг/кг/сут) была ассоциирована с плазменной экспозицией (AUC) ниже, чем у человека при МРДЧ 5 мг/сут.
Пероральное применение рисдиплама (0; 1; 3 или 7,5 мг/кг/сут) у молодых крыс от 22-го до 112-го ПНД вызывало заметное увеличение количества микроядер в костном мозге, гистопатологии мужских репродуктивных органов (дегенерация/некроз эпителия семявыносящих канальцев, олиго/аспермия в придатке яичка, гранулемы сперматозоидов) и побочные эффекты на параметры спермы (снижение концентрации и подвижности сперматозоидов, увеличение аномалий морфологии сперматозоидов) при самой высокой испытанной дозе. Увеличение количества Т-лимфоцитов (общее, хэлперы и цитотоксические) наблюдалось при средних и высоких дозах. Репродуктивные и иммунные эффекты сохранялись после прекращения применения. Доза, не оказывающая влияния (1 мг/кг/сут) на развитие побочных эффектов у молодых крыс после прекращения вскармливания, была ассоциирована с плазменной экспозицией (AUC) ниже, чем у человека при МРДЧ.
Применение в гериатрии
Клинические исследования рисдиплама не включали пациентов в возрасте 65 лет и старше для определения, отличается ли их ответ от такового у молодых взрослых пациентов.
rxlist.com, 2021.
После приема рисдиплама в виде раствора внутрь фармакокинетика была приблизительно линейной между 0.6 мг и 18 мг. Фармакокинетика рисдиплама наилучшим образом описывалась с помощью популяционной фармакокинетической модели со всасыванием с тремя транзитными камерами, двухкамерным распределением и выведением первого порядка.
Рисдиплам быстро всасывался при приеме натощак, при этом Tmax в плазме варьировало от 1 до 4 ч. Прием пищи с высоким содержанием жиров не оказывал значимого влияния на экспозицию рисдиплама. Установленные популяционные фармакокинетические параметры составили 98 л для кажущегося центрального Vd, 93 л — для периферического объема и 0.68 л/ч — для межкамерного клиренса. Рисдиплам преимущественно связывался с сывороточным альбумином без какого-либо связывания с α1-кислым гликопротеином, свободная фракция составила 11%.
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами CYP1A1, CYP2J2, CYP3A4 и CYP3A7. Фармакологически неактивный метаболит М1 был определен как основной циркулирующий метаболит.
В ходе популяционного фармакокинетического анализа был установлен кажущийся клиренс рисдиплама — 2.6 л/ч. Эффективный Т1/2 рисдиплама составил приблизительно 50 ч у пациентов со спинальной мышечной атрофией. Приблизительно 53% дозы (14% неизмененного рисдиплама) выводится с калом и 28% с мочой (8% неизмененного рисдиплама). Исходный рисдиплам был основным компонентом в плазме (83% от общих компонентов препарата, находящихся в кровотоке).
Рисдиплам
Risdiplam
Фармакологическое действие
Рисдиплам — модификатор сплайсинга мРНК, препарат для лечения мышечной атрофии позвоночника (SMA). Увеличивает системную концентрацию функциональной выживаемости белка двигательных нейронов, продуцируемого геном SMN2. Механизм действия аналогичен его предшественнику нусинерсену, наибольшее различие заключается в способе его введения: нусинерсен вводится интратекально, рисдиплам — перорально.
Рисдиплам увеличивает включение экзона 7 в транскрипты рибонуклеиновой кислоты (мРНК) SMN2 и продукцию полноразмерного белка SMN в головном мозге.
Данные in vitro и in vivo показывают, что рисдиплам может также вызывать сплайсинг других генов, включая FOXM1 и MADD, которые участвуют в регуляции клеточного цикла и апоптоза, соответственно, что может объяснять ряд неблагоприятных эффектов, наблюдаемых у животных.
Фармакодинамика
Механизм действия
Рисдиплам представляет собой модификатор сплайсинга предшественника матричной рибонуклеиновой кислоты (пре-мРНК) гена выживаемости двигательных нейронов 2 (SMN2), разработанный для лечения спинальной мышечной атрофии (СМА), причиной которой являются мутации в хромосоме 5q, что приводит к недостаточности белка SMN.
Недостаточность функционального белка SMN является патофизиологическим механизмом развития СМА всех типов. Рисдиплам корректирует сплайсинг SMN2, сдвигая баланс с исключения экзона 7 на включение экзона 7 в транскрипте м-РНК. приводя к образованию функционального и стабильного белка SMN. Таким образом, рисдиплам лечит СМА путём увеличения и сохранения уровней функционального белка SMN.
Рисдиплам равномерно распределяется во всем организме, в том числе в центральной нервной системе (ЦНС), проникая через гематоэнцефалический барьер и соответственно приводя к увеличению уровня белка SMN в ЦНС и по всему организму. Концентрации рисдиплама в плазме и белка SMN в крови отражают распределение и фармакодинамические эффекты рисдиплама в тканях, а именно: мышечных тканях и тканях мозга.
Во всех клинических исследованиях применение рисдиплама приводило к устойчивому и длительному увеличению уровня белка SMN. В течение 4 недель после начала лечения медианный уровень белка SMN был более чем в 2 раза выше по сравнению с исходным значением, согласно измерениям в крови. Повышение уровня белка SMN сохранялось на протяжении периода лечения ≤2 лет у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА (см. подраздел «Клиническая эффективность»).
Клиническая эффективность
Эффективность рисдиплама для лечения пациентов с манифестацией СМА в младенческом возрасте и пациентов с поздней манифестацией СМА оценивалось в 2 опорных клинических исследованиях FIREFISH и SUNFISH и подтверждено дополнительными данными, полученными в исследовании JEWELFISH. В целом результаты исследований подтверждают эффективность рисдиплама у пациентов со СМА.
Манифестация СМА в младенческом возрасте
ВР39056 (FIREFISH) представляет собой открытое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики рисдиплама у пациентов с симптоматической СМА 1 типа (у всех пациентов было генетически подтверждено заболевание с двумя копиями гена SMN2). Исследование состоит из двух частей. Часть 1 исследования FIREFISH разработана как часть исследования по подбору дозы. В подтверждающей части 2 исследования FIREFISH оценивалась эффективность рисдиплама в терапевтической дозе, которая была выбрана на основании результатов части 1 исследования. Пациенты из части 1 не принимали участия в части 2.
В частях 1 и 2 ключевой конечной точкой по эффективности была возможность сидеть без поддержки в течение, как минимум, 5 секунд согласно измерениям по пункту 22 шкалы развития младенцев и детей Бейли — 3 издание (Bayley Scales of Infant and Toddler Development, BSID-II1, шкала крупной моторики) после 12 месяцев лечения рисдипламом.
FIREFISH часть 2
В часть 2 исследования FIREFISH был набран 41 пациент со СМА 1 типа.
Медиана возраста возникновения клинических признаков и симптомов СМА 1 типа составила 1.5 месяцев (1.0–3.0 месяцев), 54 % были женского пола, 54 % — представители европеоидной расы и 34 % — представители азиатской расы.
Медиана возраста на момент набора в исследование составила 5.3 месяцев (2.2–6.9 месяцев), медиана времени между возникновением симптомов и приёмом первой дозы составила 3.4 месяцев (1.0–6.0 месяцев).
На исходном уровне средний индекс по результатам теста детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у новорождённых (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders, CHOP-INTEND) составил 22 балла (8.0–37.0) и средний индекс по результатам неврологической оценки младенцев по шкале Хаммерсмита, модуль 2 (Module 2 of the Hammersmith Infant Neurological Examination, HINE-2) составил 1.0 (0.0–5.0).
Первичной конечной точкой была доля пациентов с возможностью сидеть без поддержки в течение, как минимум, 5 секунд после 12 месяцев терапии (шкала крупной моторики BS1D-11I, пункт 22). Конечные точки по эффективности у пациентов, получавших рисдиплам, сравнивали с таковыми в аналогичных группах нелеченных пациентов с манифестацией СМА в младенческом возрасте с естественным течением заболевания (критерии функционирования) — см. таблицу 1 ниже.
| Конечные точки по эффективности | Доля пациентов N = 41 (90 % ДИ) |
|---|---|
|
Основные критерии двигательной функции и развития |
|
|
BSID-II1: способность сидеть без поддержки в течение, как минимум, 5 секунд Значение p на основании критерия функционирования 5 %а |
29.3 % (17.8 %, 43.1 %) <0,0001 |
|
CHOP-INTEND: индекс 40 или выше Значение p на основании критерия функционирования 17 %а |
56.1 % (42.1 %, 69.4 %) <0.0001 |
|
CHOP-INTEND: увеличение на ≥4 балла по сравнению с исходным уровнем Значение p на основании критерия функционирования 17 %a |
90.2 % (79.1 %, 96.6 %) <0.0001 |
|
HINE-2: ответившие по основным критериям двигательной функцииb Значение p на основании критерия функционирования 12 %а |
78.0 % (64,8 %, 88.0 %) <0.0001 |
|
Выживаемость и выживаемость без событий |
|
|
Выживаемость без событийc Значение p на основании критерия функционирования 42 %a |
85.4 % (73.4 %, 92.2 %) <0,0001 |
|
Живы Значение p на основании критерия функционирования 60 %a |
92.7 % (82.2 %, 97.1 %) 0.0005 |
|
Глотание и кормление |
|
|
Способность глотать |
85.4 % (73.15 %, 93.43 %) |
|
Способность принимать пищу через ротd |
82.9 % (70.3 %, 91.7 %) |
|
Использование ресурсов здравоохранения |
|
|
Отсутствие госпитализацийe |
48.8 % (35.1 %, 62.6 %) |
a Значения р по выживаемости и выживаемости без событий основаны на Z-тестировании; значения p для других конечных точек (BSID-III, CHOP-INTEND, HINE-2) основаны на точном биноминальном тестировании. Доля выживаемости устанавливается с использованием методологии Каплана-Мейера.
b Согласно HINE-2: увеличение на ≥2 балла [или максимальный показатель] для способности брыкаться, ИЛИ увеличение на ≥1 балл по основным критериям двигательной функции: способность держать голову, перекатываться, сидеть, ползать, стоять или ходить И улучшение по большему числу категорий основных критериев двигательной функции по сравнению с ухудшением, что определяется как пациент, ответивший на этот анализ.
c Явлением считается достижение конечной точки по постоянной вентиляции лёгких (трахеостомия или ≥16 часов неинвазивной вентиляции лёгких в течение суток или интубация в течение >21 последовательного дня в отсутствие или после разрешения острого обратимого явления). Три пациента достигли конечной точки по постоянной вентиляции лёгких до 12 месяца. Все 3 пациента достигли увеличения, как минимум, на 4 балла по их индексу CHOP-INTEND по сравнению с исходным уровнем.
d Включает пациентов, которых кормили исключительно через рот (всего 28 пациентов) и пациентов, которых кормили как через рот, так и с помощью трубки для кормления (всего 6 пациентов) на 12 месяце.
e Госпитализации включают все госпитализации, которые длились, как минимум, 2 дня.
После 12 месяцев терапии рисдипламом 29 % (12/41) пациентов соответствовали критерию способности сидеть без поддержки (BSID-III, пункт 22), 93 % (38/41) пациентов были живы и 85 % (35/41) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Эти результаты указывают на клинически значимое отличие от естественного течения заболевания у нелеченных пациентов с манифестацией СМА в младенческом возрасте. Нелеченные пациенты с манифестацией СМА в младенческом возрасте никогда не смогут сидеть без поддержки, и ожидается, что только 25 % пациентов смогут выжить без постоянной вентиляции лёгких после достижения возраста в 14 месяцев.
Рисунок 1. График Каплана-Мейера выживаемости без событий (FIREFISH часть 1 и часть 2)
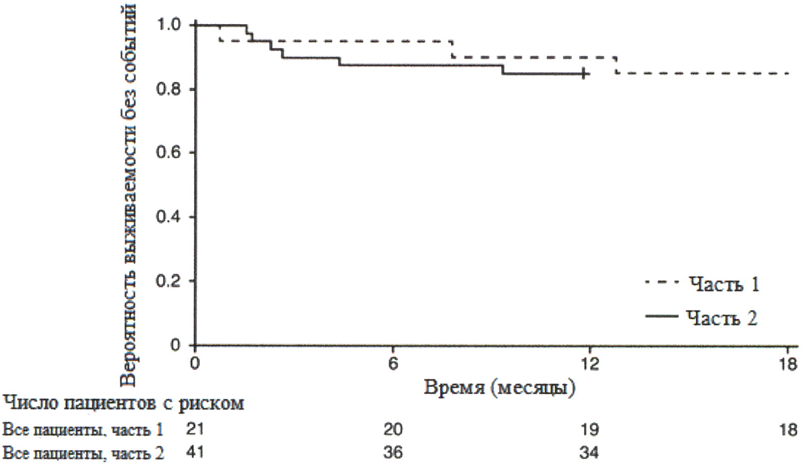
+ цензурировано: один пациент в части 2 был цензурирован, поскольку он рано осуществил визит 12 месяца.
Большинство пациентов достигли ответа по категориям основных критериев двигательной функции HINE-2, включая определённый уровень способности держать голову (76 %, 31/41), сидеть (61 %, 25/41), перекатываться (56 %, 23/41) и стоять (22 %, 9/41). Согласно измерениям общего индекса CHOP-INTEND также наблюдались улучшения двигательной функции, см. рисунок 2.
Рисунок 2. Среднее изменение по сравнению с исходным уровнем общего индекса CHOP-1NTEND (FIREFISH часть 2).

FIREFISH часть 1
Эффективность рисдиплама у пациентов со СМА 1 типа также подтверждается результатами из части 1 исследования FIREFISH. Для 21 пациента из части 1 характеристики на исходном уровне согласовывались с таковыми у пациентов с симптомами со СМА 1 типа.
Медиана возраста включения в исследование составила 6.7 месяцев (3.3–6.9 месяцев); медиана времени между возникновением симптомов и приёмом первой дозы составила 4.0 месяцев (2.0–5.8 месяцев). Всего 17 пациентов получили терапевтическую дозу (дозу, выбранную для части 2) в течение первых 12 месяцев терапии. После 12 месяцев терапии 41 % (7/17) пациентов могли сидеть без поддержки, как минимум, в течение 5 секунд (BSID-III, пункт 22). После 18 месяцев терапии 88 % (15/17) пациентов были живы и без событий (без постоянной вентиляции), см. рисунок 1. Данные результаты по выживаемости и развитию двигательной функции согласовывались с результатами исследования FIREFISH часть 2.
Поздняя манифестация СМА
ВР39055 (SUNFISH) представляет собой многоцентровое исследование по изучению эффективности, безопасности, фармакокинетики и фармакодинамики рисдиплама у пациентов со СМА 2 или 3 типа в возрасте 2–25 лет. Исследование состоит из двух частей. В части 1 осуществлялся подбор дозы. Часть 2 представляла собой рандомизированную, двойную слепую, плацебо-контролируемую, подтверждающую часть исследования. Пациенты из части 1 не принимали участия в части 2.
Первичной конечной точкой было изменение оценочного показателя двигательной функции — 32 (Motor Function Measure-32, MFM32) от исходного уровня на 12 месяце.
Индекс MFM32 позволяет оценить обширный спектр двигательных функций у большого диапазона пациентов со СМА. Общий индекс MFM32 выражают в виде процента (0-100) от максимально возможного, причём более высокий показатель обозначают лучшую двигательную функцию. Индекс MFM32 измеряет двигательную функцию, в частности важные ежедневные двигательные способности. Небольшие изменения в двигательной функции могут привести к значительному приобретению или потере ежедневных двигательных способностей.
SUNFISH часть 2
SUNFISH часть 2 представляет собой рандомизированную, двойную слепую, плацебо- контролируемую часть исследования SUNFISH у 180 не амбулаторных пациентов со СМА 2 типа (71 %) или 3 типа (29 %). Пациентов рандомизировали в соотношении 2:1 в группу лечения рисдипламом в терапевтической дозе или в группу плацебо. Рандомизация была стратифицирована по возрастным группам (2–5 лет, 6–11 лет, 12–17 лет и 18–25 лет).
Медиана возраста пациентов на момент начала лечения составляла 9,0 лет (2–25 лет), а медиана времени между возникновением первых симптомов СМА и приёмом первой дозы составила 102.6 месяцев (1–275). Из 180 пациентов, включённых в исследование, 51 % были женщинами, 67 % — представителями европеоидной расы, и 19 % — представителями азиатской расы.
На исходном уровне у 67 % пациентов выявлен сколиоз (у 32 % пациентов — тяжёлый сколиоз). Среднее значение индекса MFM32 на исходном уровне составляло 46.1, а индекса по Обновлённому модулю оценки функции верхних конечностей (Revised Upper Limb Module. RULM) — 20.1.
В целом, демографические характеристики пациентов на исходном уровне были хорошо сбалансированы между группами рисдиплама и плацебо, за исключением сколиоза (63.3% пациентов в группе рисдиплама в сравнении с 73.3% пациентов в группе плацебо).
Первичный анализ изменения общего индекса MFM32 на 12 месяце по сравнению с исходным уровнем показал клинически и статистически значимое различие между пациентами, получавшими терапию рисдипламом, и пациентами, получавшими плацебо, в исследовании SUNFISH часть 2. Результаты первичного анализа и ключевых вторичных конечных точек представлены в таблице 2 и на рисунках 3 и 4.
| Конечная точка | Рисдиплам (N = 120) |
Плацебо (N=60) |
|---|---|---|
|
Первичная конечная точка |
||
|
Изменение общего индекса MFM321 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95 % ДИ) |
1.36 (0.61, 2.11) |
-0.19 (-1.22, 0.84) |
|
Различие по сравнению с плацебо Расчётное значение (95 % ДИ) Значение р2 |
1.55 (0.30, 2.81) 0.0156 |
|
|
Вторичные конечные точки |
||
|
Доля пациентов с изменением общего индекса MFM321 на 12 месяце на ≥3 балла от исходного уровня (95 % ДИ) |
38.3 % (28.9, 47.6) |
23.7% (12.0, 35.4) |
|
Отношение шансов для общего ответа (95 % ДИ) Значение p3,4 с коррекцией (без коррекции) |
2.35 (1.01, 5.44) 0.0469 (0.0469) |
|
|
Изменение общего индекса RULM5 на 12 месяце от исходного уровня Среднее значение, рассчитанное по методу НК (95 % ДИ) |
1.61 (1.00, 2.22) |
0.02 (-0.83, 0.87) |
|
Различие по сравнению с плацебо Расчётное (95 % ДИ) значение p2,4 с коррекцией (без коррекции) |
1.59 (0.55, 2.62) 0.0469 (0.0028) |
НК = наименьшие квадраты
1 На основании правила по отсутствующим данным для индекса MFM32 6 пациентов были исключены из анализа (рисдиплам n = 115, контрольная группа плацебо n = 59).
2 Данные анализировали с использованием смешанной модели повторного измерения с общим индексом на исходном уровне, лечением, визитом, возрастной группой, лечением к визиту, а также исходным уровнем к визиту.
3 Данные анализировали с использованием логистической регрессии с общим индексом на исходном уровне, лечением и возрастной группой.
4 Значение p с коррекцией было получено для конечных точек, включённых в иерархическое тестирование, и выделялось на основании всех значений р, полученных для конечных точек в порядке увеличения иерархии до текущей конечной точки. Значение р без коррекции тестировали при уровне значимости 5%.
5 На основании правила отсутствующих данных для индекса RULM из анализа исключили 3 пациентов (рисдиплам n = 119; контрольная группа плацебо, n = 58).
При сравнении с группой плацебо пациенты, получавшие лечение рисдипламом, продемонстрировали значительное улучшение двигательной функции согласно оценке индекса MFM32 (среднее различие составило 1.55 баллов; p = 0.0156) после 12 месяцев лечения. Пациенты в возрасте 2–5 лет, получавшие терапию рисдипламом, продемонстрировали наибольшее улучшение индекса MFM32 по сравнению с контрольной группой плацебо (увеличение на ≥3 балла у 78.1 % в сравнении с 52.9 %).
Пациенты ≥18 лет, получавшие терапию рисдипламом, достигли стабилизации заболевания (изменение от исходного уровня по общему индексу MFM32 составило ≥0 баллов: 57.1 % в сравнении с 37.5 %).
У пациентов со СМА 2 типа и 3 типа, получавших терапию рисдипламом, наблюдалось сопоставимое улучшение по сравнению с исходным уровнем согласно индексу MFM32 (1.54 баллов [95 % ДИ: 0.06, 3.02]; 1.49 баллов [95 % ДИ: -0.94, 3.93] соответственно) по сравнению с контрольной группой плацебо.
Исследование также достигло вторичного независимого исхода двигательной функции, RULM. Согласно RULM отмечались статистически и клинически значимые улучшения в двигательной функции после 12 месяцев лечения по сравнению с исходным уровнем.
Пациенты в возрасте 2–5 лет, получавшие лечение рисдипламом, продемонстрировали наибольшее улучшение индекса RULM (3.41 баллов [95 % ДИ: 1.55, 5.26]), улучшение также отмечалось у пациентов ≥18 лет (1.74 баллов [95 % ДИ: -1.06, 4.53]).
Рисунок 3. Изменение среднего значения общего индекса MFM32 в течение 12 месяцев от исходного уровня (исследование SUNFISH часть 2)1
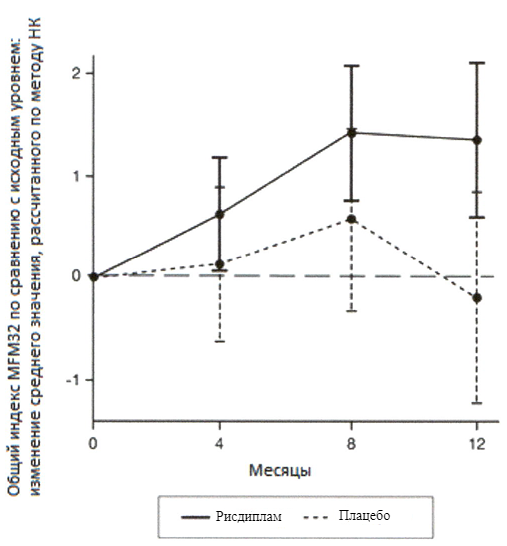
1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу MFM32 [95 % ДИ].
Рисунок 4. Изменение среднего значения общего индекса RULM в течение 12 месяцев от исходного уровня (SUNFISH часть 2)1.
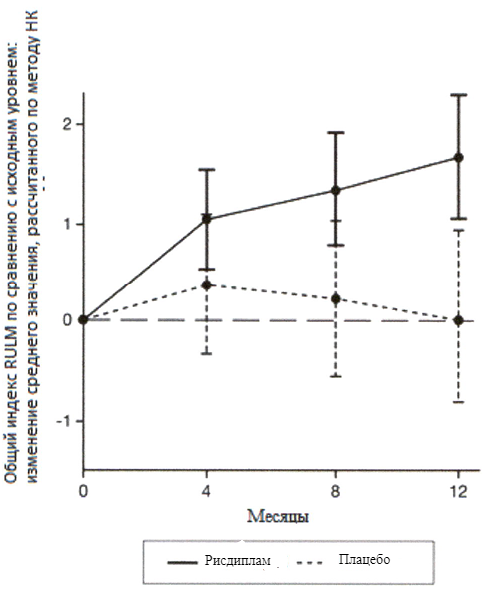
1 Отклонение среднего значения, рассчитанного по методу НК, для изменения по сравнению с исходным уровнем в баллах по индексу RULM [95 % ДИ].
SUNFISH часть 1
Эффективность рисдиплама у пациентов с поздней манифестацией СМА также подтверждается результатами из части 1 (часть исследования SUNFISH по подбору дозы).
В часть 1 был включён 51 пациент со СМА 2 типа и 3 типа (включая 7 амбулаторных пациентов) в возрасте 2–25 лет.
После 1 года лечения препаратом в терапевтической дозе (доза, выбранная для части 2) было отмечено клинически значимое улучшение двигательной функции согласно измерениям индекса MFM32. Среднее изменение по сравнению с исходным уровнем составило 2.7 баллов (95 % ДИ: 1.5, 3.8). Улучшение индекса MFM32 сохранялось ≤2 лет во время лечения рисдипламом (среднее изменение на 2.7 баллов [95 % ДИ: 1.2, 4.2]).
В поисковом анализе двигательную функцию, оцениваемую согласно индексу MFM32, в части 1 исследования SUNFISH сравнивали с группой естественного течения СМА (на основании ключевых прогностических факторов). Общее изменение индекса MFM по сравнению с исходным уровнем после 1 года и 2 лет было выше у пациентов, получавших рисдиплам, по сравнению с группой естественного течения заболевания (после 1 года: различие на 2.7 баллов; p <0.0001; после 2 лет: различие на 4.0 балла; р <0.0001). В группе естественного течения заболевания отмечалось ухудшение двигательной функции, как и ожидалось для естественного прогрессирования СМА (после 1 года: среднее изменение -0.6; после 2 лет: среднее изменение -2.0).
Применение у пациентов, ранее получавших лечение по поводу СМА
ВР39054 (JEWELFISH) представляет собой одногрупповое, открытое исследование по изучению безопасности, переносимости, фармакокинетики и фармакодинамики рисдиплама у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. В исследование были включены пациенты в возрасте 6 месяцев — 60 лет, которые ранее получали лечение по поводу СМА (включая нусинерсен и онасемноген абепарвовек). Из 174 пациентов, включённых в исследование, 76 пациентов ранее получали терапию нусинерсеном (9 пациентов со СМА 1 типа, 43 пациента со СМА 2 типа и 24 пациента со СМА 3 типа) и 14 пациентов ранее получали онасемноген абепарвовек (4 пациента со СМА 1 типа и 10 пациентов со СМА 2 типа). После 4 недель терапии рисдипламом у пациентов отмечалось увеличение уровня белка SMN в крови в среднем ≥2 раза выше по сравнению с исходным уровнем.
Фармакокинетика
Фармакокинетические параметры рисдиплама были изучены у здоровых взрослых добровольцев и у пациентов со СМА.
После приёма рисдиплама в виде раствора внутрь фармакокинетика рисдиплама была приблизительно линейной между 0.6 и 18 мг. Фармакокинетика рисдиплама наилучшим образом описывалась с помощью популяционной фармакокинетической модели со всасыванием с тремя транзитными камерами, двухкамерным распределением и выведением первого порядка. Было обнаружено, что масса тела и возраст оказывают значимое влияние на фармакокинетику.
При применении рисдиплама в терапевтической дозе 0.2 мг/кг один раз в сутки у пациентов с манифестацией СМА в младенческом возрасте (от 2 до 7 месяцев при наборе в исследование) расчётная экспозиция (средняя площадь под кривой «концентрация-время» (AUC)0-24 ч) составила 1930 нг.ч/мл. В исследовании SUNFISH (часть 2) при применении рисдиплама в терапевтической дозе 0.25 мг/кг один раз в сутки у пациентов с массой тела <20 кг и 5 мг один раз в сутки у пациентов с массой тела >20 кг (пациенты с поздней манифестацией СМА от 2 до 25 лет при наборе в исследование), расчётная экспозиция составила 2070 нг.ч/мл. Наблюдаемая максимальная концентрация (средняя Cmax) составила 194 нг.ч/мл при применении в дозе 0.2 мг/кг в исследовании FIREFISH и 120 нг.ч/мл в исследовании SUNFISH часть 2.
Всасывание
Рисдиплам быстро всасывался при приёме натощак, при этом время достижения максимальной концентрации (tmax) в плазме варьировало от 1 до 4 часов после приёма внутрь. Приём пищи (высококалорийный завтрак с высоким содержанием жиров) не оказывал значимого влияния на экспозицию рисдиплама.
Распределение
Установленные популяционные фармакокинетические параметры составили 98 л для кажущегося центрального объёма распределения, 93 л для периферического объёма и 0.68 л/ч для межкамерного клиренса.
Рисдиплам преимущественно связывался с сывороточным альбумином без какого-либо связывания с альфа-1-кислым гликопротеином, свободная фракция составила 11 %.
Метаболизм
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1A1, 2J2, ЗA4 и ЗA7.
Одновременный приём итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приёмом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11 %, показатель Cmax снижен на 9 %).
Выведение
В ходе популяционного фармакокинетического анализа был установлен кажущийся клиренс (CL/F) рисдиплама 2.6 л/ч.
Эффективный период полувыведения рисдиплама составил приблизительно 50 часов у пациентов со СМА.
Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Приблизительно 53 % дозы (14 % неизменного рисдиплама) выводилось с калом и 28 % с мочой (8 % неизменного рисдиплама). Исходный препарат был основным компонентом в плазме, что составило 83 % от общих компонентов препарата, находящихся в кровотоке.
Фармакологически неактивный метаболит М1 был определён как основной циркулирующий метаболит.
Особые группы пациентов
Пациенты детского возраста
Масса тела и возраст были определены как ковариаты в популяционном фармакокинетическом анализе. Таким образом, доза определяется в зависимости от возраста (младше и старше 2 лет) и массы тела (до 20 кг) для достижения сходных экспозиций среди пациентов всех возрастов и с различной массой тела. Данные у пациентов младше 2 месяцев отсутствуют.
Пациенты пожилого возраста
Специальных исследований по изучению фармакокинетики рисдиплама у пациентов со СМА старше 60 лет не проводилось. Пациенты со СМА ≤60 лет принимали участие в исследовании JEWELFISH. Субъекты без СМА ≤69 лет принимали участие в клинических исследованиях по фармакокинетике. Согласно результатам данных исследований коррекции дозы у пациентов ≤69 лет не требуется.
Пациенты с нарушением функции почек
Исследований по изучению фармакокинетики рисдиплама у пациентов с нарушением функции почек не проводилось. Рисдиплам в виде неизменного соединения выводится почками в малой степени (8 %).
Пациенты с нарушением функции печени
Нарушение функции печени лёгкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. После применения 5 мг рисдиплама средние соотношения для Cmax и AUC составили 0.95 и 0.80 у субъектов с нарушением функции печени лёгкой степени тяжести (n = 8), а также 1.20 и 1.08 у субъектов с нарушением функции печени средней степени тяжести (n =  по сравнению с соответствующими здоровыми добровольцами (n = 10). Безопасность и фармакокинетика у пациентов с нарушением функции печени тяжёлой степени тяжести не изучались.
по сравнению с соответствующими здоровыми добровольцами (n = 10). Безопасность и фармакокинетика у пациентов с нарушением функции печени тяжёлой степени тяжести не изучались.
Этническая принадлежность
Фармакокинетика рисдиплама у японских субъектов и субъектов европеоидной расы не отличалась.
Показания
Спинальная мышечная атрофия у взрослых и детей в возрасте от 2 месяцев и старше.
Противопоказания
Повышенная чувствительность к рисдипламу, беременность, лактация, возраст младше 2 месяцев.
Беременность и грудное вскармливание
Применение при беременности
Категория действия на плод по FDA — N.
Адекватных и хорошо контролируемых исследований о возможности применения рисдиплама у беременных женщин не проведено.
Исследования на животных выявили неблагоприятные последствия (эмбриофетальная смертность, пороки развития, снижение массы тела плода и репродуктивные нарушения у потомства).
У женщин детородного возраста до начала терапии должна быть исключена возможная беременность.
Женщины с репродуктивным потенциалом должны использовать эффективные средства контрацепции во время лечения и в течение как минимум 1 месяца после последней дозы препарата.
Рисдиплам противопоказан при беременности.
Применение в период грудного вскармливания
Специальных исследований о возможности применения рисдиплама в период грудного вскармливания не проведено.
Неизвестно, выделяется ли рисдиплам в грудное человеческое молоко. Экспериментальные исследования показали выделение рисдиплама в молоко лактирующих животных. Риск для грудного ребёнка не может быть исключён.
Фертильность
Лечение рисдипламом может снизить мужскую фертильность.
Рекомендуется предупреждать пациентов мужского пола о потенциальных неблагоприятных репродуктивных эффектах (возможно рассмотрение вопроса о сохранении спермы перед началом лечения). Пациенты-мужчины, которые хотят завести ребёнка, должны прекратить лечение препаратом, как минимум, на 4 месяца. Лечение может быть продолжено после зачатия.
На основании данных доклинических исследований влияния рисдиплама на фертильность у женщин не ожидается.
Контрацепция
Пациенты (мужчины и женщины) репродуктивного потенциала должны соблюдать следующие требования в отношении контрацепции:
- пациенты-женщины детородного потенциала должны использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приёма последней дозы;
- пациенты-мужчины и их партнёрши детородного потенциала должны вместе использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 4 месяцев после приёма последней дозы пациентом-мужчиной.
Способ применения и дозы
Внутрь 1 раз в день после приёма пищи. Рекомендуемая доза для взрослых и детей массой тела более 20 кг — 5 мг в сутки; для детей массой тела менее 20 кг — 0,2–0,25 мг/кг веса.
Лечение СМА следует начать как можно раньше после постановки диагноза.
Побочные действия
Резюме профиля безопасности
Профиль безопасности рисдиплама основан на трёх клинических исследованиях FIREFISH, SUNFISH и JEWELFISH.
FIREFISH представляет собой открытое исследование, состоящее из двух частей, в которое включено 62 пациента с манифестацией СМА в младенческом возрасте (от 2.2 до 6.9 месяцев). Пятьдесят пять пациентов получали терапию рисдипламом в течение >12 месяцев (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»). Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с манифестацией СМА в младенческом возрасте, представленные в таблице 5 ниже, основаны на данных объединённого анализа у пациентов из частей 1 и 2 клинического исследования FIREFISH. Нежелательные реакции определяются как нежелательные явления, которые отмечаются у ≥5% пациентов и для которых возможна причинно-следственная связь с рисдипламом.
SUNFISH представляет собой исследование, состоящее из двух частей, в которое включены пациенты с поздней манифестацией СМА (от 2 до 25 лет) (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Нежелательные реакции, которые наблюдались в клинических исследованиях у пациентов с поздней манифестацией СМА, основаны на данных исследования SUNFISH часть 2 (n = 180), рандомизированной, двойной слепой, плацебо-контролируемой части исследования с периодом последующего наблюдения, как минимум, 12 месяцев.
Нежелательные реакции определяются как нежелательные явления, которые отмечаются на ≥5% чаще или, как минимум, в 2 раза чаще по сравнению с пациентами, получавшими плацебо, и для которых возможна причинно-следственная связь с рисдипламом.
| Класс систем органов | Нежелательная реакция | Частота N = 62 n (%) |
Число явлений/ 100 пациенто-лет Общая экспозиция пациенто-лет = 87.9 |
Категория частоты |
|---|---|---|---|---|
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 10 (16.1) | 13.7 | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 17(27.4) | 23.9 | Очень часто |
* Включая сыпь, макулопапулёзную сыпь, эритему, дерматит, аллергический дерматит, папулёзную сыпь, фолликулит.
| Класс систем органов | Нежелательная реакция | Частота N = 120 n (%) |
Плацебо N = 60 n (%) |
Категория частоты |
|---|---|---|---|---|
| Нарушения со стороны желудочно-кишечного тракта | Диарея | 20 (16.7) | 5 (8.3 %) | Очень часто |
| Нарушения со стороны кожи и подкожных тканей | Сыпь* | 20 (16.7) | 1 (1-7) | Очень часто |
Включая сыпь, макулопапулёзную сыпь, эритему, аллергический дерматит, эритематозную сыпь, фолликулит, папулёзную сыпь.
Нежелательные реакции диарея и сыпь отмечались без определённого времени или клинической картины и разрешались несмотря на продолжающееся лечение рисдипламом у пациентов с манифестацией СМА в младенческом возрасте и у пациентов с поздней манифестацией СМА. Данные явления не указывают на влияние на эпителиальные ткани, которые отмечались в исследованиях у животных.
Профиль безопасности у пациентов, ранее получавших терапию по поводу СМА
Профиль безопасности рисдиплама у пациентов, ранее получавших терапию по поводу СМА в исследовании JEWELFISH, сопоставим с таковым у пациентов, ранее не получавших лечения в исследованиях FIREFISH (часть;= 1 и;= 2) и SUNFISH (часть;= 1 и;= 2). В исследовании JEWELFISH принимали участие 76;= пациентов, которые ранее получали нусинерсен и;= 14;= пациентов, которые ранее получали онасемноген абепарвовек (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Передозировка
Случаев передозировки в клинических исследованиях не было.
Лечение
Известного антидота для применения в случае передозировки рисдиплама нет.
В случае передозировки следует тщательно наблюдать за пациентом и провести поддерживающую терапию.
Взаимодействие
Рисдиплам в основном метаболизируется флавин-монооксигеназой 1 и 3 (FMO 1 и 3), а также изоферментами цитохрома (CYP) 1A1, 2J2, ЗA4 и ЗA7. Рисдиплам не является субстратом белка множественной лекарственной устойчивости 1 (MDR1) у человека.
Влияние других лекарственных средств на рисдиплам
Одновременный приём итраконазола, мощного ингибитора CYP3A, в дозе 200 мг два раза в сутки с однократным пероральным приёмом рисдиплама в дозе 6 мг не выявил клинически значимого влияния на фармакокинетику рисдиплама (показатель AUC повышен на 11 %, показатель Cmax снижен на 9 %). При одновременном применении ингибитора CYP3A и рисдиплама коррекции дозы последнего не требуется.
Лекарственного взаимодействия с препаратами, метаболизирующимися посредством флавин-монооксигеназы 1 и 3 (FMO 1 и 3), не ожидается.
Влияние рисдиплама на другие лекарственные средства
В условиях in vitro рисдиплам и его основной циркулирующий метаболит М1 не индуцировали CYP1A2, 2B6, 2C8, 2C9, 2C19 или ЗA4. В условиях in vitro рисдиплам и M1 не ингибировали (обратимо или в зависимости от времени) какой-либо из изучаемых ферментов CYP (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6) за исключением CYP3A.
Рисдиплам является слабым ингибитором CYP3A. При введении препарата рисдиплам здоровым взрослым добровольцам один раз в сутки в течение двух недель экспозиция мидазолама (чувствительного субстрата CYP3A) незначительно повышалась (AUC 11 %, Cmax 16 %). Степень взаимодействия не считается клинически значимой, и, таким образом, коррекции дозы субстратов CYP3A не требуется. Согласно фармакокинетическому моделированию, основанному на физиологии, ожидается, что данный эффект будет иметь сходную выраженность у младенцев старше 2 месяцев и детей.
Исследования in vitro показали, что рисдиплам и его основной метаболит не являются значимыми ингибиторами MDR1, транспортного полипептида органических анионов OATP1B1 и OATP1BЗ и переносчика органических анионов OAT 1 и OAT3 у человека.
Рисдиплам и его метаболит, однако, являются ингибиторами переносчика органических катионов ОСТ2, белка экструзии лекарственных препаратов и токсинов MATE1 и MATE2-K в условиях in vitro. При терапевтических концентрациях препарата взаимодействий с субстратами ОСТ2 не ожидается. Согласно данным, полученным в условиях in vitro, при применении рисдиплама плазменные концентрации препаратов, которые выводятся посредством MATE1 и MATE2-K, могут повышаться.
Клиническая значимость одновременного применения с субстратами MATE1/2-K неизвестна.
Особые указания
Эмбриофетальная токсичность
При изучении применения препарата у животных наблюдалась эмбриофетальная токсичность. Пациенты репродуктивного потенциала должны быть проинформированы о рисках и использовать высокоэффективные методы контрацепции во время лечения и в течение, как минимум, 1 месяца после приёма последней дозы рисдиплама пациентом-женщиной и в течение, как минимум, 4 месяцев после приёма последней дозы рисдиплама пациентом-мужчиной (см. раздел «Беременность и грудное вскармливание»).
Возможное влияние на фертильность у мужчин
Обратимое влияние рисдиплама на мужскую фертильность было показано в исследовании у животных. В связи с этим, пациенты-мужчины не должны быть донорами спермы во время лечения и в течение 4 месяцев после приёма последней дозы рисдиплама (см. раздел «Беременность и грудное вскармливание»).
Нарушение функции печени
Фармакокинетику, безопасность и переносимость рисдиплама в дозе 5 мг оценивали у субъектов с нарушением функции печени лёгкой или средней степени тяжести в специальном клиническом исследовании.
Нарушение функции печени лёгкой или средней степени тяжести не оказывало влияния на фармакокинетику рисдиплама. Таким образом, пациентам с нарушением функции печени лёгкой или средней степени тяжести коррекции дозы не требуется. Применение рисдиплам у пациентов с нарушением функции печени тяжёлой степени тяжести не изучалось (см. раздел «Фармакологические свойства», подраздел «Особые группы пациентов».
Злоупотребление приёмом препарата или лекарственная зависимость
Рисдиплам не обладает потенциалом, который может привести к злоупотреблению приёмом препарата и лекарственной зависимости.
Влияние на способность к вождению автотранспорта и управлению механизмами
Препарат не оказывает влияния на способность управлять транспортными средствами, механизмами.
Классификация
-
АТХ
M09AX10
-
Фармакологическая группа
-
Коды МКБ 10
-
Категория при беременности по FDA
N
(не классифицировано FDA)
Информация о действующем веществе Рисдиплам предназначена для медицинских и фармацевтических специалистов, исключительно в справочных целях. Инструкция не предназначена для замены профессиональной медицинской консультации, диагностики или лечения. Содержащаяся здесь информация может меняться с течением времени. Наиболее точные сведения о применении препаратов, содержащих активное вещество Рисдиплам, содержатся в инструкции производителя, прилагаемой к упаковке.

Коротко
- «Дженентек» (Genentech), входящая в состав «Рош» (Roche), раскрыла годовые результаты продолжающихся опорных клинических исследований FIREFISH (NCT02913482) фазы II/III (нерандомизированных, открытых, многоцентровых), проверяющих экспериментальный рисдиплам (risdiplam) в терапии спинальной мышечной атрофии типа I среди пациентов в возрасте 1–7 месяцев с двумя копиями гена SMN2. Ранее были опубликованы полугодовые результаты лечения. Кроме того, представлены промежуточные результаты терапии спинальной мышечной атрофии типов II и III.
- Спинальная мышечная атрофия вызывается гомозиготной делецией или мутацией гена выживаемости мотонейронов 1 (SMN1), кодирующего белок SMN, который жизненно необходим для нормального развития и функционального гомеостаза и который экспрессирует на нейрональных и других клетках. У большинства людей есть также родственный ген SMN2, способный продуцировать белок SMN. Всё бы ничего, но ген SMN2, ввиду трансляционно синонимичной мутации С>T в экзонном энхансере сплайсинга, кодирует пре-мРНК, которая, проходя альтернативный сплайсинг, исключает экзон 7 из большинства мРНК-транскриптов. Результирующая мРНК SMN2Δ7 продуцирует белок SMNΔ7, но он, будучи нестабильным, быстро распадается. При этом только 10–20% мРНК-транскриптов SMN2 становятся полноразмерными белками SMN, чего критически недостаточно.
- Поскольку у всех пациентов со спинальной мышечной атрофией (без функционального гена SMN1) есть хотя бы одна копия SMN2 (а у некоторых их число доходит до шести в соматических клетках), имеет смысл организовать альтернативный сплайсинг пре-мРНК гена SMN2 путем включения ошибочно исключенного экзона 7.
Подробности
Рисдиплам, разрабатываемый «Рош» совместно с «ПиТиСи терапьютикс» (PTC Therapeutics), — низкомолекулярное соединение, которое высокоизбирательно организует указанную процедуру. Лекарственное соединение, принимаемое ежедневно перорально, минует гематоэнцефалический барьер и системно распределяется по организму. Отправка регистрационного досье регуляторам намечена на второе полугодие.
В настоящее время «Спинраза» (Spinraza, нусинерсен), за которым стоят «Байоджен» (Biogen) и «Айонис фармасьютикалс» (Ionis Pharmaceuticals), является единственным доступным препаратом против спинальной мышечной атрофии. Давеча регуляторное разрешение получил «Золгенсма» (Zolgensma, онасемноген абепарвовек) — генная терапия авторства «Авексис» (AveXis) и «Новартис» (Novartis), обещающая фактически исцеление заболевания (ну или выход к долгосрочной ремиссии) после одной инъекции, однако за сумасшедшие деньги.

По прошествии года лечения рисдипламом 58,8% пациентов (n=10/17, группа терапевтической дозы) со спинальной мышечной атрофией типа I достигли 40 и более баллов по шкале младенческих нейромышечных заболеваний теста Детской больницы Филадельфии (CHOP-INTEND), причем максимум зафиксировался на 57 баллах. Медианное увеличение получилось равным 17,5 баллов.
Для оценки тенденций улучшения двигательных навыков уместно напомнить, как улучшался балл CHOP-INTEND по ходу терапии. Так, после двух месяцев назначения рисдиплама медианная прибавка составила 5,5 балла (n=20/21), после трех — 12,5 балла (n=16/21), после шести — 14 баллов (n=11/21).
Во время терапии, медианная продолжительность которой составила 14,8 месяца, три младенца столкнулись с фатальными осложнениями заболевания после приблизительно одного, восьми и 13 месяцев лечения. Впрочем, ни одно из таковых не связано с назначением рисдиплама. Никто из пациентов не потерял возможность глотания, никому не потребовалась трахеостомия или постоянная искусственная вентиляция легких. Бессобытийная выживаемость вышла к 85,7% (n=18/21) и 88,2% (n=15/17) — соответственно для всей популяции больных и в группе с терапевтической дозой.
По прошествии 12 месяцев приема рисдиплама 41,2% респондентов (n=7/17) отметились способностью сидеть без посторонней помощи в течение минимум пяти секунд, 64,7% (n=11/17) — сидеть (с поддержкой или без нее), 52,9% (n=9/17) — удержания головы в вертикальном положении, 5,9% (n=1/16) — стоять без поддержки. Следует понимать, что при естественном течении спинальной мышечной атрофии типа I, оставляемой без какого-либо лечения, указанных нейромышечных навыков не наблюдается в принципе.
 «Дженентек» также поделилась промежуточными результатами опорных клинических испытаний SUNFISH (NCT02908685) фазы II/III (рандомизированных, двойных слепых, плацебоконтролируемых), изучающих рисдиплам среди пациентов со спинальной мышечной атрофией типов II и III. Исходные показатели участников варьируются в широких пределах: от неспособности сидеть до умения ходить.
«Дженентек» также поделилась промежуточными результатами опорных клинических испытаний SUNFISH (NCT02908685) фазы II/III (рандомизированных, двойных слепых, плацебоконтролируемых), изучающих рисдиплам среди пациентов со спинальной мышечной атрофией типов II и III. Исходные показатели участников варьируются в широких пределах: от неспособности сидеть до умения ходить.
Поисковый анализ эффективности, выполненный для 43 пациентов из 51, установил, что для 58% (включая 71% среди больных в возрасте 2–11 лет и 42% — 12–25 лет) справедливо улучшение минимум на три балла согласно шкале оценки моторных функций MFM32.
 Окончательные итоги FIREFISH и SUNFISH будут подведены в первом квартале 2020 года и четвертом квартале 2019-го. Ну а регистрационная заявка на рисдиплам запланирована к отправке во второй половине текущего года.
Окончательные итоги FIREFISH и SUNFISH будут подведены в первом квартале 2020 года и четвертом квартале 2019-го. Ну а регистрационная заявка на рисдиплам запланирована к отправке во второй половине текущего года.
Параллельно идут клинические испытания JEWELFISH (NCT03032172) и RAINBOWFISH (NCT03779334), в которых рисдиплам тестируется соответственно среди пациентов в возрасте от шести месяцев до 60 лет с любым типом спинальной мышечной атрофии, прежде лечившихся каким-либо SMN-таргетным препаратом или олезоксимом (olesoxime), и среди предсимптоматических больных, заболевание которых заблаговременно выявлено генетическим тестированием.
Важная информация
Mosmedpreparaty.ru — специализированная научно-исследовательская и справочно-информационная аналитическая служба группы компаний «Мосмедпрепараты», таргетированная на ключевые события глобальной отрасли фармации, биотехнологий, медицины и здравоохранения.
- Ничто на Mosmedpreparaty.ru не является рекламой или продвижением лекарственных препаратов, методов лечения, медицинских услуг.
- Сведения и публикации Mosmedpreparaty.ru носят исключительно научно-просветительский и ознакомительный характер.
- Медицинская информация, транслируемая Mosmedpreparaty.ru, предназначена только для специалистов в области здравоохранения и сфере обращения лекарственных средств.
- Медицинская информация, содержащаяся на Mosmedpreparaty.ru, не предназначена для использования в качестве замены консультации со специалистом в области здравоохранения.
- Ничто на Mosmedpreparaty.ru не должно истолковываться как предоставление медицинского совета или рекомендации и не может служить основанием для принятия каких-либо решений или осуществления каких-либо действий без участия специалиста в области здравоохранения.
Присутствие на веб-ресурсе Mosmedpreparaty.ru и ознакомление с его содержимым означает, что вы прочитали «Пользовательское соглашение» и приняли его условия.