РУКОВОДСТВО
ПО МЕТОДАМ ОПРЕДЕЛЕНИЯ ВРЕДНЫХ ВЕЩЕСТВ
В АТМОСФЕРНОМ ВОЗДУХЕ
РЕФЕРАТ
Санитарная охрана атмосферного воздуха приобретает особо важное значение в связи с широким развитием промышленности и автотранспорта в нашей стране. Органы санитарного контроля изучают состояние воздушной среды населенных мест, устанавливая предельно допустимые концентрации вредных веществ в воздухе. Для успешного проведения подобных исследований необходимо располагать объективными и надежными методами определения вредных веществ в атмосферном воздухе. Чем точнее полученные данные, тем эффективнее будут проведены оздоровительные мероприятия по охране атмосферного воздуха от загрязнений.
Настоящая книга является пособием по методам определения вредных веществ в атмосферном воздухе и состоит из 15 глав. В главе I изложены общие вопросы по определению атмосферных загрязнений: даны указания по отбору проб, максимально разовых и среднесуточных, подробно описаны способы отбора проб воздуха, приборы и аппаратура, применяемые для этой цели, способы приготовления стандартных растворов, формулы расчета анализа, приведения объема воздуха к нормальным условиям и т.д.
В 15 главах руководства описаны методы определения токсических веществ, распределенных по классам соединений, например соединения серы, соединения азота, металлы, аминосоединения и др.
В книге описаны методы определения отдельных токсических веществ в атмосферном воздухе. Методы составлены по единой схеме, перед изложением каждого метода приведены физико-химические и токсические свойства определяемого соединения, а также предельно допустимые концентрации — максимальные разовые и среднесуточные. Для ряда токсических веществ дано несколько методов, что расширяет возможности определения этих веществ при различных условиях.
Приведенные в руководстве методы позволяют в первую очередь определять вещества, на которые установлены предельно допустимые концентрации. Кроме того, приводится описание методов определения веществ, вновь синтезируемых и внедряемых в промышленность и являющихся источником загрязнения атмосферного воздуха.
Настоящее руководство рассчитано на химиков, работающих в санитарно-эпидемиологических станциях, лабораториях гидрометеорологической службы, а также в ведомственных лабораториях.
ПРЕДИСЛОВИЕ
Охрана внешней среды, в том числе атмосферного воздуха, в нашей стране поставлена на уровень важнейших государственных задач.
Решения XXIV съезда КПСС по усилению охраны природы в стране, Основы законодательства Союза ССР и союзных республик о здравоохранении (утверждены VII сессией Верховного Совета СССР 19 декабря 1969 г.), Постановление «О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов» (принято 20 сентября 1972 г.) целеустремленно направлены на бережное отношение к природе, в частности, на охрану чистоты атмосферного воздуха как важнейшего фактора, обеспечивающего состояние здоровья населения.
Указанные постановления партии и правительства обязывают руководителей министерств, ведомств, предприятий, учреждений, проектных и строительных организаций предусматривать в перспективных и годовых планах проведение природооздоровительных мероприятий по предупреждению загрязнения атмосферного воздуха, водоемов, подземных вод и почвы и претворять их в жизнь.
Вместе с тем рост промышленности и транспорта все еще неизбежно ведет к загрязнению атмосферы городов и других населенных пунктов. Основными источниками загрязнения воздушного бассейна вредными веществами являются ТЭЦ, котельные, транспорт, различные двигатели внутреннего сгорания, а также предприятия металлургической и химической промышленности. Следует отметить, что в связи с химизацией народного хозяйства значительно увеличился выброс в атмосферу различных соединений сложного состава, ранее не известных и не безопасных для здоровья населения.
В успешной борьбе за чистоту атмосферного воздуха существенное место занимают санитарный надзор и дальнейшие, вытекающие из обстановки гигиенические мероприятия.
Систематический санитарно-лабораторный контроль в стране осуществляют санитарно-эпидемиологические станции, лаборатории гидрометеорологической службы и в соответствии с Постановлением ЦК КПСС и Совета Министров СССР N 517 от 1968 г. ведомственные и производственные лаборатории.
Санитарный надзор по охране атмосферного воздуха является эффективным в нашей стране благодаря разработанным критериям оценки вредности различных веществ, базирующимся на основе материалистического учения И.П. Павлова. Принципы гигиенического нормирования вредных веществ в атмосферном воздухе основаны на фундаментальных исследованиях советских гигиенистов.
Гигиенические нормативы лежат в основе санитарного законодательства и позволяют получать объективные данные о степени и опасности загрязнения воздушной среды в населенных местах. В этих условиях, естественно, трудно переоценить значение объективных, надежных методов лабораторного контроля воздушной среды.
Чем точнее получены данные, тем эффективнее будут оздоровительные мероприятия по охране атмосферного воздуха от загрязнения. Следует подчеркнуть особую сложность разработки методических решений в области охраны атмосферного воздуха. С одной стороны, необходимо разработать методы, позволяющие определить микроколичества веществ, с другой — следует учитывать, что в атмосферном воздухе всегда одновременно присутствуют группы и целые комплексы различных веществ, осложняющие аналитическую работу.
Настоящая книга является фундаментальным пособием по методам определения вредных веществ в атмосферном воздухе.
Книга рассчитана главным образом на химиков, работающих в санитарно-эпидемиологических станциях, а также на работников различных ведомственных учреждений, выполняющих анализы воздушной среды.
В основу книги положен большой многолетний опыт работы авторов в области санитарной химии в Московском научно-исследовательском институте гигиены имени Ф.Ф. Эрисмана и в других научно-исследовательских институтах гигиенического профиля, также использован литературный материал.
В книге дается краткая характеристика физико-химических свойств групп веществ, приводится их токсичность, описаны наиболее точные и одновременно общедоступные методы определения этих веществ в атмосферном воздухе.
Изложение книги начинается с основ лабораторной практики, что весьма необходимо, особенно начинающим химикам.
Разработаны рекомендации по отбору максимально разовых и среднесуточных проб воздуха, имеющих принципиальное значение при гигиенической оценке воздушной среды и атмосферных загрязнений.
Подробно изложены основы современных методических исследований с учетом использования новой лабораторной техники (фотометрии, хроматографии и др.). Одновременно даны вполне доступные методы исследования для небольших лабораторий.
Директор
Московского научно-исследовательского
института гигиены имени Ф.Ф. Эрисмана
профессор
член-корреспондент АМН СССР
А.П.ШИЦКОВА
ВВЕДЕНИЕ
Советская медицина стоит на позициях предупреждения и своевременного устранения различных факторов внешней среды, оказывающих вредное влияние на здоровье человека.
В решениях XXIV съезда КПСС, в Основах законодательства Союза ССР и союзных республик, а также в Постановлении Верховного Совета СССР «О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов» задачи защиты чистоты атмосферного воздуха и предохранения его от загрязнения признаны общенародными, общегосударственными, от правильного и своевременного решения которых во многом зависит состояние здоровья населения нашей страны [11, 22, 24, 26].
Для защиты воздушного бассейна от загрязнения промышленными выбросами устанавливают мощные очистительные сооружения, газо-пылевые устройства, проводят строительство поясов зеленых насаждений вокруг городов и промышленных центров, а также проводят другие санитарно-гигиенические мероприятия.
К сожалению, отдельные очистные сооружения работают пока еще недостаточно эффективно, иногда имеют место непредусмотренные выбросы, аварийные случаи, что влечет за собой загрязнение атмосферного воздуха населенных мест. Это свидетельствует о техническом несовершенстве отдельных очистительных сооружений.
Особенно сильно загрязняет воздушный бассейн автомобильный транспорт, выхлопные газы которого с содержащимися в них токсическими соединениями находятся в прямой зависимости от режима работы двигателей.
Дальнейшее развитие химической, нефтехимической промышленности, а также черной и цветной металлургии и увеличивающееся использование двигателей внутреннего сгорания связано с повышенным количеством выбрасываемых веществ в атмосферный воздух. При этом не только растет число разнообразных химических веществ, выбрасываемых в атмосферный воздух, но и усложняется состав выбросов. В свою очередь наличие токсических веществ в атмосферном воздухе является источником как дискомфорта, так и прямой угрозы здоровью населения.
Токсические вещества имеют широкий диапазон вредного воздействия. Так, в одних случаях они вызывают токсикозы, в других создают неблагоприятные условия для произрастания и формирования растений, вызывают коррозию строительных материалов и др.
Влияние вредных атмосферных загрязнений на здоровье человека имеет свою специфику, с которой следует считаться. Дело в том, что в производственных помещениях, воздух которых загрязнен токсическими соединениями, как правило, работают взрослые. Пребывание их в помещениях, атмосферный воздух которых загрязнен токсическими соединениями, ограничивается только рабочим временем. Другое дело, когда речь идет о загрязнении атмосферного воздуха населенного пункта. В этом случае он воздействует непрерывно и днем, и ночью, в течение всего времени загрязнения воздушного бассейна и, что самое главное, его воздействию подвергаются люди всех возрастов — от самого раннего до старческого, которые особенно чувствительны к токсическим веществам.
Все это и заставляет работников здравоохранения изучать и решать вопросы, связанные с охраной атмосферного воздуха. Вполне понятно, что решить эту проблему можно лишь совместными усилиями технологов, гигиенистов, химиков, физиологов, физиков, токсикологов и специалистов других профессий.
Роль химиков в этом велика и ответственна, они определяют, какими веществами загрязнен воздух и в каком количестве. От правильного заключения будет зависеть и направленность всех проводимых оздоровительных мероприятий.
Практически контроль за состоянием воздушного бассейна в нашей стране осуществляют органы здравоохранения. В их ведении и подчинении находятся специальные лаборатории Министерства здравоохранения СССР. Кроме того, работы по исследованию атмосферного воздуха проводятся и в лабораториях ведомственных учреждений. Большую работу по исследованию воздуха ведут гидрометеорологические службы страны; проводятся крупные научно-исследовательские, гигиенические работы, связанные с анализом атмосферного воздуха.
Во исполнение Постановления ЦК КПСС и Совета Министров СССР от 5 июля 1968 г. N 517 «О мерах по дальнейшему улучшению здравоохранения и развития медицинской науки в стране» на промышленных предприятиях должна быть организована еще более широкая сеть лабораторий по осуществлению постоянного контроля за соблюдением санитарно-гигиенических нормативов, в частности за состоянием атмосферного воздуха.
Естественно, что для успешного проведения таких работ необходимо располагать методами исследования атмосферного воздуха. В данном руководстве изложены методы определения большого числа токсических веществ в атмосферном воздухе. Почти по каждому веществу представлен ряд методов, используемых в зависимости от условий.
В руководстве в основном изложены методы, используемые при исследовании атмосферного воздуха, однако отдельные высокочувствительные и проверенные на практике методы заимствованы из области исследования воздуха производственных помещений.
В настоящее время в практике исследования атмосферных загрязнений широко используются фотометрические методы, реже — хроматографические, полярографические и другие виды анализа.
Сущность отдельных видов анализа в руководстве не освещена потому, что более подробное ее описание приводится в специальной литературе.
В данном руководстве в первую очередь описаны те токсические соединения, на которые утверждены предельно допустимые концентрации (ПДК) в воздухе населенных мест.
В общей части руководства изложены некоторые основные положения, связанные с определением малых количеств токсических веществ. В специальном разделе даются методы отбора проб и определения токсических веществ в атмосферном воздухе. Методы определения отдельных токсических веществ описаны по единой схеме.
Перед изложением методов определения того или иного соединения дана краткая информация о физических и токсикологических свойствах, приведены предельно допустимые, максимально разовые и среднесуточные концентрации.
Далее описаны принципы метода, указана чувствительность в определяемом объеме или в 1 мл пробы. Также указано влияние некоторых примесей, проверенных авторами методов. Приведен список необходимых реактивов, описан отбор проб, изложен ход анализа.
Возможно, что некоторые вопросы остались неосвещенными, поэтому авторы будут благодарны за критические замечания и дополнения по данному руководству.
Глава I
ОБЩИЕ ВОПРОСЫ ПО ОПРЕДЕЛЕНИЮ АТМОСФЕРНЫХ ЗАГРЯЗНЕНИЙ
Атмосферный воздух представляет собой смесь газов: азота, кислорода, углекислого газа, аргона, неона и др. Состав атмосферного воздуха до высоты 85 — 100 км практически одинаков, в составе околоземных слоев сухого атмосферного воздуха присутствуют следующие компоненты (табл. 1).
Таблица 1
СОСТАВ СУХОГО АТМОСФЕРНОГО ВОЗДУХА [3]
|
Компоненты |
Процент по весу |
Компоненты |
Процент по весу |
|
Азот |
75,50 |
Гелий |
0,00007 |
|
Кислород |
23,12 |
Водород |
0,00005 |
|
Аргон |
1,285 |
Криптон |
0,0000025 |
|
Углекислый газ |
0,046 |
Радон |
6 · 10-18 |
|
Неон |
0,001 |
Содержание озона у поверхности земли находится в пределах 3 · 10-6 по объему. Концентрация его увеличивается с высотой и достигает максимума на уровне 25 — 30 км.
В воздухе содержится 0 — 4% водяных паров по объему.
В связи с ростом металлургической, химической и других видов промышленности атмосферный воздух загрязняется многими токсическими веществами.
В основном источником загрязнения воздушного бассейна являются промышленные предприятия, размещенные без соблюдения санитарно-гигиенических норм и правил или эксплуатирующиеся с их нарушением.
Количество токсических веществ, поступающих в атмосферный воздух от действующих предприятий, зависит от ряда обстоятельств, в том числе от объема и количества выбросов, условий поступлений их в атмосферу. Концентрация токсических веществ меняется в зависимости от высоты труб, систематичности и случайности выбросов, от удаления точек, где проводят исследование, от источника загрязнения, ландшафта местности, направления и силы ветра, градиента температуры, влажности и др.
Атмосферный воздух загрязняется также топочными газами котельных и выхлопными газами автотранспорта.
В атмосферном воздухе под действием ионизирующих излучений происходят фотохимические процессы с образованием окислов азота, озона и т.д. Озон образуется при облучении воздуха ультрафиолетовой радиацией с длиной волны 253 нм, а окислы азота — до 100 нм. Поверхности земли достигает радиация лишь с длиной волны около 290 нм.
В условиях загрязнения атмосферного воздуха промышленными выбросами, выхлопными газами автотранспорта фотохимические реакции проходят под действием обычной солнечной радиации. При этом возможно превращение окиси азота в двуокись, накопление озона в атмосфере и др. При взаимодействии углеводородов с озоном или атомарным кислородом образуются свободные пероксильные, высокореактивные вещества, способные вступать в реакцию с окислами азота и другими соединениями и образовывать сложный комплекс веществ, обладающих окислительными свойствами, — оксиданты.
При особых метеорологических условиях, способствующих накоплению оксидантов, в атмосферном воздухе может образовываться фотохимический смог, сопровождающийся уменьшением прозрачности (снижением дальности видимости), появлением неприятных запахов, раздражением слизистых оболочек глаз, носа, горла, увяданием растительности и др. С целью обнаружения токсических веществ, находящихся в виде газов, паров, аэрозолей, пыли, проводятся гигиенические исследования атмосферных загрязнений. Обычно воздух загрязнен токсическими веществами, находящимися в весьма сложном сочетании, что в ряде случаев затрудняет проведение анализа.
Санитарный контроль за чистотой атмосферного воздуха осуществляется путем отбора проб с анализом этих проб.
Исследуя атмосферный воздух как с научной, так и с практической целью, приходится выполнять анализы проб, в которых содержатся весьма малые количества веществ. Это вызвано высокой степенью разбавления выбросов в атмосфере и низкими ПДК для воздуха населенных мест [25].
Это указывает на специфические трудности, возникающие при проведении анализа атмосферных загрязнений, и налагает особую ответственность на химиков-аналитиков при выборе наиболее чувствительных и по возможности избирательных методов исследования.
Исследования атмосферного воздуха связаны с определением микрограммовых количеств веществ, и понятно, что при анализе воздушной среды должны применяться высокочувствительные методы, а сам анализ должен быть выполнен в строгом соответствии с правилами, предъявляемыми к аналитической химии малых концентраций.
При анализах воздуха высокочувствительными методами необходимо учитывать, что если определяемая величина оказывается близкой к чувствительности метода, то ошибка определения может быть весьма ощутимой. Во избежание этого следует, например, применяя колориметрические методы, использовать по возможности калибровочный график или сравнивать интенсивность окраски со шкалой в средней части графика или шкалы [19, 20].
При определении микроколичеств следует учитывать также наличие сопутствующих веществ, иногда превышающих искомое вещество, а также фон, который образуется постоянными или случайными загрязнениями, попадающими из реактивов, посуды и т.п.
Применяемая посуда должна быть тщательно промыта и пропарена. Приспособление для пропаривания показано на рис. 1.
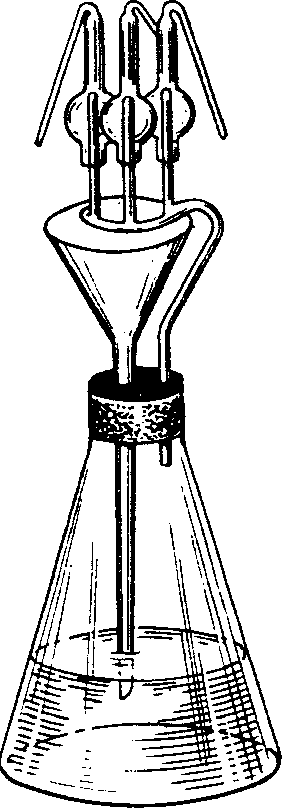
Рис. 1. Приспособление для пропаривания
При проведении анализов проб атмосферного воздуха необходимо устранять возможные потери, происходящие за счет улетучивания, адсорбции, ионного обмена; частицы золы могут быть унесены конвенционными потоками и др. Следует обращать внимание и на потери, которые могут быть при применении некачественной фарфоровой посуды (трещины, вплавление в стенки сосуда и др.). Во избежание этого необходимо тщательно соблюдать режим нагревания, не допускать разбрызгивания и соблюдать другие предосторожности. С целью, например, устранения потерь искомого вещества при фильтровании можно пользоваться малой массой фильтровальной бумаги. Для этого делают соскоб с фильтра и получают волокна. Фильтрование ведут в небольшой воронке с оттянутым концом (рис. 2), в который вкладывают тампон из волокон фильтра длиной 0,2 — 0,3 см (вес около 0,05 г). Если возможно по ходу анализа, волокна в воронке предварительно промывают разбавленным раствором кислоты.
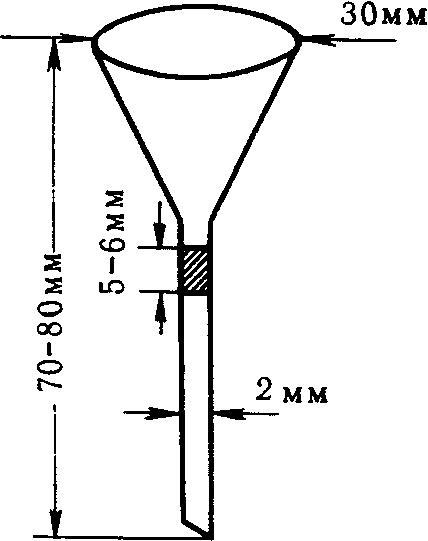
Рис. 2. Воронка для фильтрования
С целью нивелирования погрешности анализа обязательно подготавливают контрольное определение, проведенное в идентичных с анализом пробы условиях. Повышение чувствительности определения достигается различными приемами. Применяется способ концентрирования исследуемого вещества в процессе отбора проб, что осуществляется увеличением скорости протягивания воздуха в единицу времени или увеличением времени отбора пробы. При применении жидких поглотительных сред указанный прием возможен лишь при получении в процессе поглощения нелетучего продукта.
Достигается это и в процессе анализа концентрированием пробы, упариванием, отгонкой, экстрагированием, хроматографированием, уменьшением растворимости осадка; применяются приборы с большей разрешающей способностью, используются специальные приспособления для кювет и др. [7, 9].
Некоторые методы достаточно чувствительны и не требуют накопления веществ, что позволяет проводить определение в малом объеме (до 1 л). При этом отбирают пробы воздуха в газовые пипетки.
Определение токсических веществ в атмосферном воздухе складывается из двух основных разделов — отбора проб и последующего анализа. Оба раздела имеют самостоятельное значение, дополняют друг друга, и качество отбора проб влияет на результат определения. Критерием оценки загрязнения являются данные, полученные в результате отбора и анализа максимальных разовых и среднесуточных концентраций вредных веществ в атмосферном воздухе [1, 6].
ОТБОР ПРОБ ВОЗДУХА [14]
Постоянный контроль за состоянием атмосферного воздуха осуществляется путем отбора максимальных разовых и среднесуточных проб воздуха в точках, расположенных в заранее определяемых местах. С целью правильного размещения точек отбора проб необходимо располагать планом исследуемого района с нанесением на этот план основных ориентиров. Следует иметь данные, характеризующие то или иное предприятие как источник загрязнения атмосферного воздуха. Это поможет установить время отбора проб воздуха в течение суток или сезона, а также определить расстояния, на которых необходимо вести отбор проб воздуха.
При расстановке точек отбора проб учитывают метеорологические и рельефные особенности размещения производств, сложившуюся ситуацию по взаиморазмещению объектов и жилых районов, а также другие специфические особенности.
Одновременно с отбором проб на всех точках производят наблюдения за скоростью и направлением ветра, отмечают атмосферное давление и температуру.
Отбор максимальных разовых проб. При изучении загрязнений атмосферного воздуха проводят отбор проб в зонах максимального загрязнения, непосредственно в факеле выброса. Отбор проб может быть проведен последовательно в каждой зоне обязательно с захватом факела.
Лучшим вариантом отбора максимальных разовых проб следует считать одновременный отбор во всех обследуемых зонах, причем в 2 — 4 точках, расположенных поперек в каждой зоне факела. Расстояние крайних точек относительно осевой обычно не превышает 5% расстояния от источника выброса до зоны исследования (например, в зоне 2000 м 2 точки должны быть расставлены на расстоянии не более 100 м от точки, находящейся на оси). При таком отборе проб учитывают максимальные показатели, боковые точки лишь подтверждают правильность расстановки точек.
Отбор проб под факелом осуществляется, как правило, в течение 20 мин. и не превышает 30 мин. В каждой точке отбирают не менее 25 проб в течение ряда дней на уровне 1,5 м от земли (в зоне дыхания). При большом разбросе полученных данных эта величина может оказаться недостаточной.
В отдельных случаях отбирают разовые пробы в стационарах или подвижных точках без учета факела. В этих случаях отбор проб лучше проводить то в утренние, то в вечерние часы суток, в часы максимального загрязнения воздуха. Отбирают пробы на уровне 1,5 м от земли (в зоне дыхания), в местах, где нет пыли, например на газоне, твердом грунте и т.п. Отбор проб в подвижных и стационарных точках обычно не превышает 30 мин.
Отбор проб проводят аспирированием исследуемого воздуха через поглотительный прибор, в который помещен тот или иной сорбент, накапливают токсическое вещество до необходимых пределов. В отдельных случаях отбор проб проводят за короткий промежуток времени, например в газовые пипетки. При этом с целью получения средних данных по концентрации токсических веществ за 20 — 30 мин. можно отбирать последовательно 5 — 6 проб с одинаковыми интервалами.
Отбор среднесуточных проб. Среднесуточные пробы отбирают на стационарных и подвижных точках, в зоне дыхания человека на высоте 1,5 м от земли, на открытых площадках в удалении от строений. Выбирают точки отбора там, где нет пыли. В стационарных точках отбор проб проводят периодически, не меняя их расположения. В подвижных точках отбор проб проводят также в течение суток, но, в зависимости от изменяющихся условий, перемещают точки отбора.
При отборе проб в стационарных точках не учитывают направление ветра, и, если пункт наблюдения выходит из факела, отбор проб продолжают.
Обычно для отбора проб выбирают период наибольшего загрязнения атмосферного воздуха по условиям выброса или метеорологическим данным. Желательно размещать как можно больше точек отбора, но если такой возможности нет, то при отборе проб в передвижных точках следует в разные дни выезда менять их расположение.
Рекомендуется отбирать среднесуточные пробы в количестве не менее 10 — 15 в сезон. Пробы отбирают в течение суток, причем допускается несколько вариантов исследований:
1) исследуемый воздух протягивают непрерывно в течение суток через один и тот же поглотительный прибор, наполненный поглотительным раствором, через фильтр, помещенный в патрон, или твердый сорбент;
2) исследуемый воздух протягивают через один и тот же поглотительный прибор, наполненный поглотительным раствором, через фильтр или твердый сорбент, с перерывами в определенные промежутки времени, через 2 или 4 ч (без смены поглотительного прибора, патрона). Отбирают пробы в течение суток 6 или 12 раз по 20 — 30 мин.;
3) в случае необходимости нужно проследить за динамикой загрязнения воздуха района, в различные часы суток можно использовать способ отбора проб в разные поглотительные приборы, т.е. провести отбор проб, например, 6, 12 или 24 раза через одинаковые промежутки времени, при этом воздух протягивают в течение 20 — 30 мин. и каждую пробу анализируют отдельно. При таком способе удается установить периоды максимального и минимального загрязнения.
При отборе проб воздуха необходимо учитывать состояние определяемого вещества в воздухе (газ, пар, аэрозоли).
В зависимости от этого отбор проб осуществляют в поглотительные приборы, содержащие поглотительные среды, или на фильтрующие материалы, помещенные в патроне.
Для поглощения токсических веществ из воздуха применяют аспирационные способы или способы отбора проб в сосуды ограниченной емкости.
Аспирационные способы отбора проб. Отбор проб атмосферного воздуха проводят в населенных пунктах и в полевых условиях, вдали от населенных мест и от производства. В зависимости от условий применяют различные аспирационные устройства. Используют аспираторы, работающие от электросети или аккумуляторов, применяют автомобильный или водяные аспираторы различных конструкций.
Поглощение газов и паров в жидкие поглотительные среды. К жидким поглотительным средам относятся дистиллированная вода, органические растворители, специальные поглотительные растворы.
Поглощение газов и паров может происходить путем растворения их в жидкой среде или в результате взаимодействия токсического вещества с поглотительным раствором. Если поглощение основано на растворении вещества в растворителе, то такое поглощение может быть отнесено к малоэффективным. Обычно поглощение токсического вещества происходит при малых скоростях протягивания исследуемого воздуха и интенсивном охлаждении.
Увеличение скорости отбора проб, а также повышение температуры ведет к улетучиванию как растворителя, так и исследуемого вещества. В таком случае отбор среднесуточных проб возможен с перерывами 6 — 24 раза в отдельные поглотительные приборы с индивидуальным анализом (третий вариант). Если поглощение токсических веществ проводят в поглотительные растворы, содержащие соответствующие реактивы, то в процессе поглощения происходит реакция взаимодействия его с реактивом, обычно с образованием нелетучего соединения, чем обеспечивается более полное поглощение.
Следует учитывать, что во время отбора проб, особенно среднесуточных, происходит испарение растворителя, при этом объем поглотительной среды уменьшается. Во избежание изменения объема пробы периодически в процессе отбора пробы объем поглотительной жидкости доводят до первоначального уровня растворителем, используемым при анализе.
Эффективность поглощения вещества достигается возможно большим контактом его с поглощающей жидкой средой. С этой целью употребляются поглотительные приборы различных конструкций.
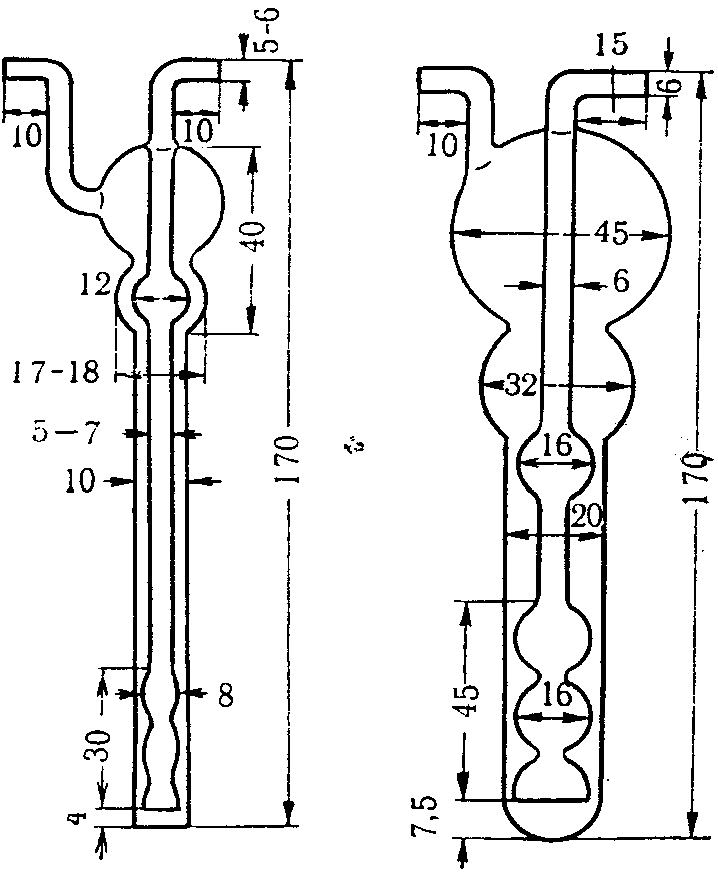
Поглотительные приборы с пористой стеклянной пластинкой (рис. 3) представляют собой V-образные трубки с впаянными в виде пластин стеклянными фильтрами N 1 и 2. Стеклянные фильтры сделаны из особо приготовленной спекшейся массы стекла с различными величинами пор.
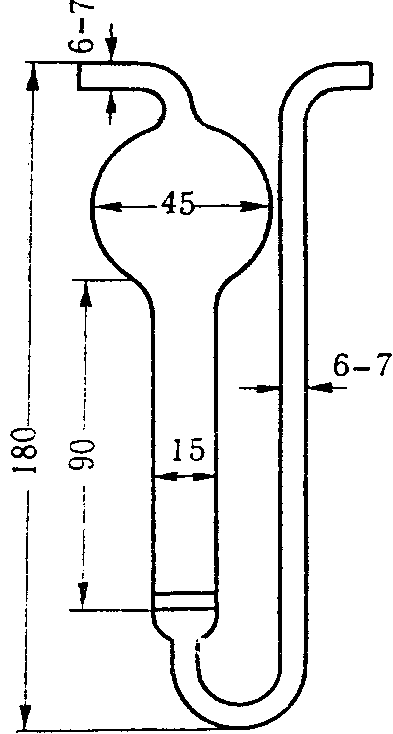
Рис. 3. Поглотительный прибор
с пористым стеклянным фильтром
N 1 — ПС-1 диаметр пор 90 — 150 мк, N 2 — ПС-2 диаметр пор 40 — 90 мк. Сопротивление фильтров устанавливают, наливая воду на фильтр высотой 5 мм, и протягивают воздух до появления первых пузырьков. Для ПС-1 сопротивление должно быть 25 — 15 мм рт. ст., для ПС-2 55 — 25 мм рт. ст. Диаметр фильтров 15 и 10 мм, толщина 2 — 3 мм (ГОСТ 9775-61). Используются поглотительные приборы емкостью 2 и 6 мл (малая и большая модель, диаметр пористой пластинки от 10 мм и более). В поглотительный прибор вносят жидкость, через которую протягивают исследуемый воздух. Воздух при помощи впаянной стеклянной пористой пластинки разбивается на множество мелких пузырьков, чем обеспечивается большая поверхность соприкосновения с поглотительной средой. Поглотительные приборы, снабженные пористой пластинкой, относятся к эффективным, обеспечивающим интенсивное поглощение, позволяющим проводить отбор проб со скоростью до 2 — 3 л/мин.
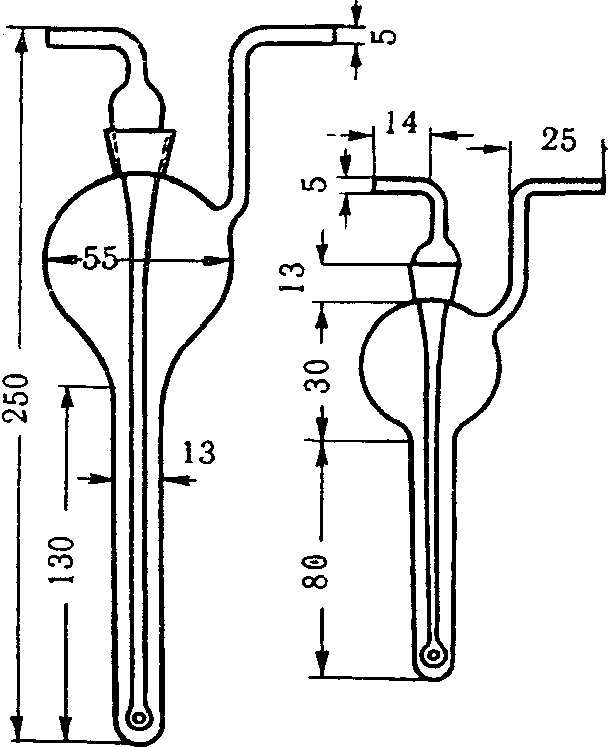
Поглотительные приборы Зайцева, Полежаева, Рыхтера (рис. 4) представляют собой стеклянные сосуды, внутри которых проходит цилиндрическая трубка, переходящая в некоторых случаях в расширение. В поглотительные сосуды вносят жидкую поглотительную среду по 2 — 10 мл. При применении поглотительных приборов Зайцева воздух протягивают со скоростью до 1 л/мин.; поглотительные приборы Полежаева обеспечивают поглощение при скорости протягивания воздуха до 0,5 л/мин., а приборы Рыхтера — до 10 л/мин.
Поглотительный прибор Гернет (рис. 5) представляет собой стеклянный грушевидной формы сосуд с распылителем типа форсунки. Воздух поступает в распылитель со скоростью 10 — 15 л/мин. При этом поглотительная среда распыляется внутри прибора и достигается большая поверхность соприкосновения жидкой среды с воздухом. Выводится воздух из прибора через верхнюю трубку, снабженную ловушкой со стеклянными бусами, служащими для улавливания брызг.
А
Б
В
Рис. 4. Поглотительные приборы Зайцева (А),
Рыхтера (Б), Полежаева (В)
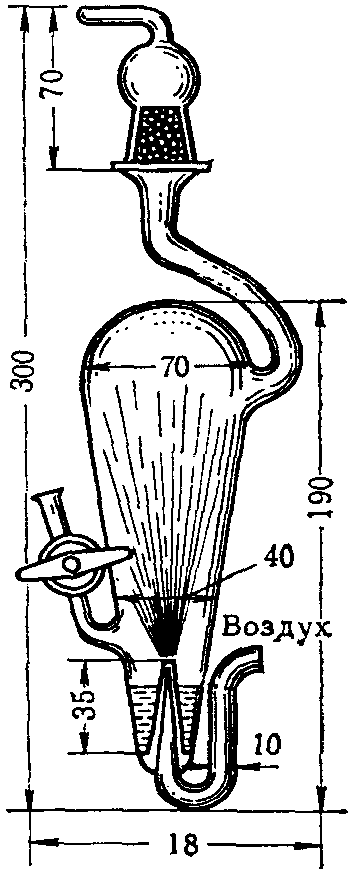
Рис. 5. Поглотительный прибор Гернет
Поглощение газов и паров твердыми зерненными сорбентами [15, 16]. К твердым поглотительным средам относят зерненные сорбенты. При исследовании атмосферного воздуха чаще всего используют силикагель и активные угли. Твердые зерненные сорбенты характеризуются рядом положительных свойств — они удобны при хранении и транспортировке, обладают повышенными сорбционными свойствами при пониженных температурах.
Силикагель — пористое тело с сильно развитой поверхностью, зерна его характеризуются механической прочностью. Силикагель обладает ценным качеством — способностью сорбировать вещества и после специальной обработки восстанавливать свои первоначальные свойства. В зависимости от величины пор и формы зерен силикагель разделяют на мелкопористый, крупнопористый, кусковой и гранулированный.
В гигиенической практике обычно используют кусковой (как мелкопористый, так и крупнопористый) силикагель. Некоторые сведения, касающиеся кускового силикагеля, приведены в табл. 2 <1>.
———————————
<1> 1 Гранулированный силикагель содержит упрочняющую зерна добавку — 4 — 10% алюминия (ГОСТ 3956-54, силикагель).
Таблица 2
КУСКОВОЙ СИЛИКАГЕЛЬ
|
Марки силикагеля |
Величина зерен, мм |
|
КСМ — крупный силикагель мелкопористый |
2,7 — 7 |
|
ШСМ — шихта силикагель мелкопористый |
1,5 — 3,5 |
|
МСМ — мелкий силикагель мелкопористый |
0,25 — 3,5 |
|
АСМ — активированный силикагель мелкопористый |
0,2 — 0,5 |
|
КСК — крупный силикагель крупнопористый |
2,7 — 7 |
|
ШСК — шихта силикагель крупнопористый |
1,5 — 3,5 |
|
МСК — мелкий силикагель крупнопористый |
0,25 — 3,5 |
|
АСК — активированный силикагель крупнопористый |
0,2 — 0,5 |
Для поглощения токсических веществ чаще всего применяют силикагель с величиной частиц 0,25 — 2,00 мм, поэтому крупный силикагель, например КСМ, ШСМ, МСМ, КСК, ШСК, МСК, размельчают в ступке и, применяя соответствующий размер сита, готовят зерна необходимой величины.
Если силикагель окрашен (обычно за счет содержания железа), то его обрабатывают горячей соляной кислотой (удельный вес 1,19) и далее 10% раствором соляной кислоты до получения неокрашенного раствора. Затем силикагель промывают водой до удаления иона хлора (по реакции с азотнокислым серебром), высушивают при 100 °С и активируют при 200 — 150 °C в течение часа в сушильном шкафу. Хранят силикагель в закрытых склянках.
Силикагель перед употреблением проверяют на отсутствие вещества, подлежащего определению.
Температура в пределах +/- 40° и относительная влажность в интервалах 50 — 85% практически не влияют на сорбционную способность силикагеля.
В ряде случаев для поглощения токсических веществ применяют активные угли. Уголь, как и силикагель, представляет собой пористое тело, состоящее из углерода. Угли типа Г, АГ-5, АГ-3 активные, гранулированные, газовые, поглощающие пары и газы вследствие сильно развитой удельной поверхности. Уголь, как и силикагель, перед употреблением проверяют на отсутствие вещества, которое определяют, т.е. проводят контрольный опыт.
Для сорбции токсических веществ на твердых зерненных сорбентах последние помещают в специальные трубки или поглотительные приборы. Поглощение веществ проводят в неподвижном или «кипящем» слое.
При поглощении токсического вещества на неподвижный слой сорбент помещают в трубки различных конфигураций, иногда с перемежающимися сферическими расширениями (рис. 6). Сорбент в количестве до 5 см3 помещают в трубки, в концы которых закладывают стеклянную вату или спирали из проволоки для закрепления зерен. Ввиду большого сопротивления в таких трубках до 300 мм вод. ст. исследуемый воздух протягивают со скоростью лишь до 2 л/мин. и применяют мощные аспирационные устройства.
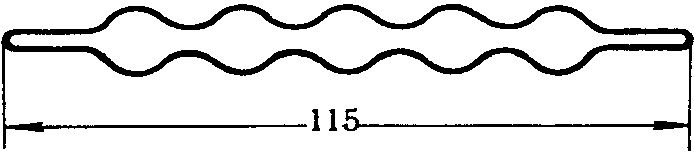
Рис. 6. Трубка со сферическими расширениями
для твердого сорбента
Поглощение токсических веществ из атмосферного воздуха можно проводить на кипящий слой зерненного сорбента.
При этом целесообразно использовать силикагель, так как его зерна обладают механической прочностью. Зерна силикагеля помещают в поглотительный прибор и через слой протягивают воздух. По мере увеличения скорости подачи воздуха, а следовательно, и его объема через слой сорбента этот слой значительно увеличивается, твердые частицы перестают касаться друг друга и приобретают подвижность. Образуется кипящий слой, обусловливающий эффективность перемешивания и контакт с исследуемым веществом. Отбор проб проводят со скоростью до 12 л/мин., при этом сопротивление кипящего слоя сравнительно невелико (60 мм вод. ст.).
Для отбора проб воздуха на кипящий слой используются поглотительные приборы Яворовской и Московского научно-исследовательского института гигиены имени Ф.Ф. Эрисмана и др. (рис. 7). Через боковую трубку поглотительных приборов при помощи стеклянной воронки вводят до 5 мл зернистого сорбента. С целью устранения переброса частиц силикагеля в поглотительных приборах предусмотрены вогнутые вглубь шипы.
Переброс зерен страхуется последовательным присоединением специального прибора, показанного на рис. 7. По окончании отбора проб переброшенные частицы силикагеля присоединяются к общей массе. Токсические вещества, поглощенные на сорбент, десорбируют путем экстрагирования соответствующими растворителями или растворами, в отдельных случаях десорбцию веществ осуществляют выдуванием с последующим поглощением в соответствующий раствор.
Поглощение аэрозолей. Для поглощения аэрозолей из воздуха используют фильтрующие материалы: плотные бумажные фильтры (беззольные), фильтры из тонких волокон и др., которые в виде дисков помещают в специальные патроны. Бумажные фильтры, обладая задерживающей способностью, оказывают большое сопротивление. Беззольный бумажный фильтр (синяя лента) с рабочей поверхностью 18 кв. см при скорости протягивания воздуха до 10 л/мин. имеет сопротивление до 300 мм вод. ст.
Рис. 7. Поглотительные приборы для отбора проб в кипящий
слой Московского научно-исследовательского института имени
Ф.Ф. Эрисмана (А), Яворовской (Б), для задерживания
переброшенных частиц (В)
В практике исследования воздушной среды для поглощения аэрозолей широко используются аналитические фильтры аэрозольные (АФА).
Фильтры АФА обладают высокой задерживающей способностью и практически полностью задерживают аэрозоли размером 0,1 — 0,2 мк при скорости протягивания до 100 л/мин. Фильтр обладает небольшим собственным весом, негигроскопичен (гидрофобен), стоек к химически агрессивным средам, растворим в ацетоне, дихлорэтане; пары и газообразные примеси фильтр АФА не задерживает. Фильтры АФА обладают сравнительно небольшим сопротивлением, например фильтр с рабочей поверхностью 18 кв. см имеет сопротивление 20 — 40 мм вод. ст. при скорости протягивания воздуха 20 л/мин.
Фильтры АФА хранят в закрытом виде при температуре не выше 40 °C в сухом помещении, в отсутствие паров масел, ацетона, дихлорэтана.
Осуществляется массовый выпуск фильтров АФА-В-10, АФА-В-18 для весового анализа с рабочей поверхностью 10 и 18 кв. см, АФА-XII-18 — перхлорвиниловые, предназначенные для химического анализа, и др. [8, 28].
Патроны представляют собой воронку, в широкой части которой при помощи специального кольца герметизируется фильтр. Ширина выреза в патроне соответствует рабочей поверхности фильтра. На рис. 8 указаны патроны и фильтры, используемые для отбора проб на аэрозоли; на рис. 9 показан патрон, используемый для задержания сажи. В отдельных случаях для поглощения аэрозолей применяют фильтры, предназначенные для отбора проб пыли, имеющие большую рабочую поверхность.
Рис. 8. Фильтр и патрон для отбора проб аэрозолей
А — общий вид и основные элементы фильтра;
Б — патрон для фильтров, имеющих рабочую
поверхность 18 и 20 кв. см
Рис. 9. Патрон для задерживания сажи
(рабочая поверхность 22 кв. мм)
Аспирационные приборы
Отбор проб в жидкие, твердые поглотительные среды, а также на фильтры проводят динамическим аспирационным способом при помощи специальных приборов. Такими приборами являются водяные аспираторы, электроаспираторы. Применяют и другие аспирационные устройства. Например, в практике широко используются автомобильные аспираторы, в отдельных случаях пользуются баллонами со сжатым воздухом, водоструйным насосом и т.д.
При этом объем исследуемого воздуха определяют при помощи самих водяных аспираторов или с применением измерительной аппаратуры.
Простейшими приборами для отбора проб воздуха являются водяные аспираторы, работающие по принципу сообщающихся сосудов.
Объем вытекаемой воды соответствует количеству воздуха, протянутого через поглотительный прибор (рис. 10).
Отбор круглосуточных проб проводится водяным аспиратором системы Рязанова (рис. 11). Этот аспиратор состоит из двух баков, расположенных один под другим. Емкость баков до 500 л. Баки сообщаются между собой через трубку с краном, которым регулируется скорость вытекания воды из верхнего бака в нижний.
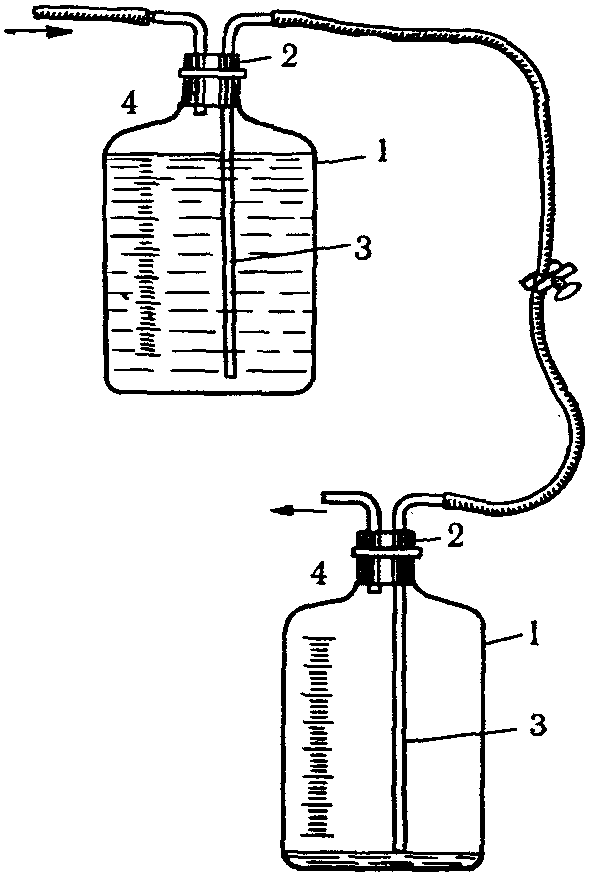
Рис. 10. Водяной аспиратор, изготовленный из двух бутылей
1 — бутыль; 2 — пробка; 3 — длинная трубка — сифон;
4 — короткая трубка
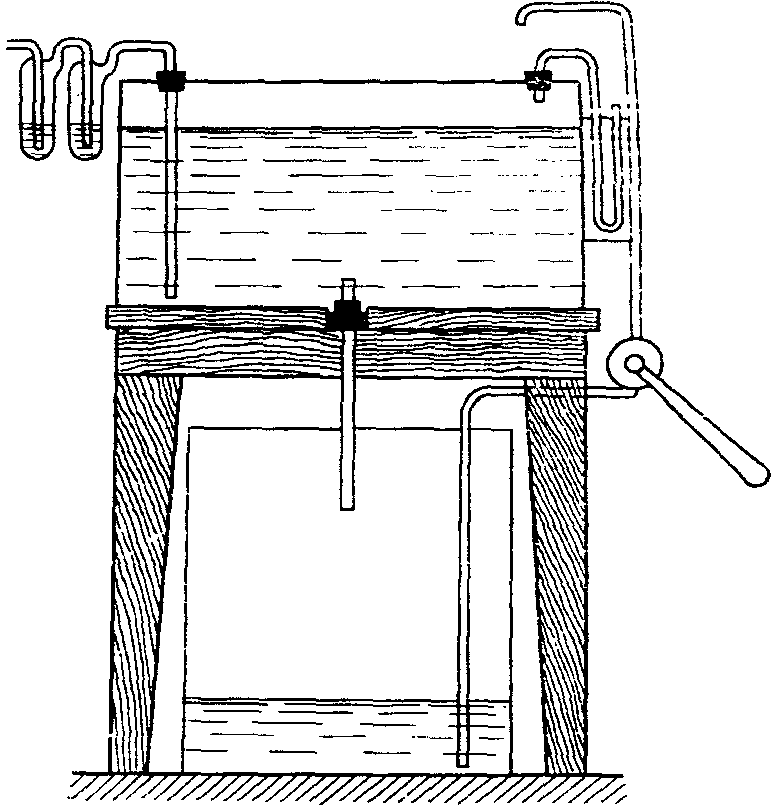
Рис. 11. Водяной аспиратор системы Рязанова
Отбор проб проводят со скоростью до 500 л в сутки, около 10 — 20 л в час. Зимой аспиратор наполняют 26% раствором поваренной соли.
Отбор проб можно проводить, используя автомобильный двигатель как аспиратор, а в зимнее время как средство обогрева поглотительных приборов, заполненных жидкой поглотительной смесью.
Для отбора проб предложено специальное приспособление к санитарной автомашине УАЗ-450а, ПАЗ-653 [10].
Аспирация исследуемого воздуха осуществляется при помощи всасывающего коллектора двигателя и шланга. К шлангу присоединена съемная панель, на которой размещены системы, состоящие из реометров и поглотительных приборов (рис. 12). При работе двигателя аспирируется воздух со скоростью от 1 до 100 л/мин.
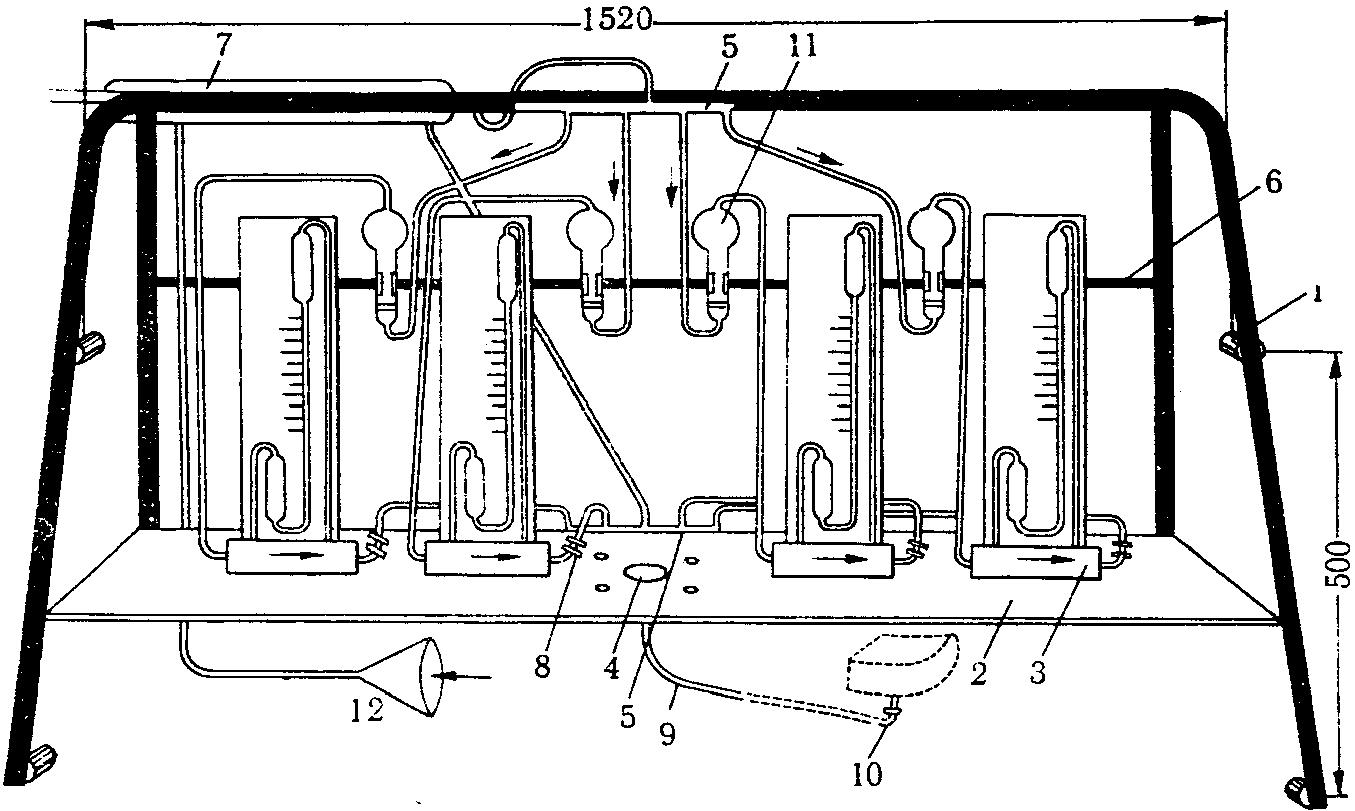
Рис. 12. Схема съемной приборной панели
1 — место крепления; 2 — полка для реометров; 3 — реометры;
4 — место крепления; 5 — гребенка; 6 — крепление
поглотительных приборов; 7 — 13 — место крепления
теплообменника; 8 — зажимы; 9 — шланги; 10 — всасывающий
коллектор двигателя; 11 — поглотительные приборы;
12 — воронка, через которую поступает исследуемый воздух
Для отбора проб воздуха широко применяют электроаспираторы, работающие от электролиний; вдали от электросети используются электроаспираторы, работающие на аккумуляторах.
Для отбора проб используют и бытовые пылесосы различной конструкции, при этом удобно пользоваться специальной приставкой [27].
На рис. 13 схематически изображена приставка и дан общий вид установки. Приставка представляет собой дюралюминиевую трубку с расширением в центральной части, где имеется 8 патрубков одинакового диаметра (1). Воздух протягивают со скоростью до 10 л/мин. С одной стороны трубки присоединен еще один патрубок большего диаметра (2), соединенный с винтом (3), которым регулируется степень разрежения в патрубках. Вращением винта прикрывают величину отверстия патрубка (4) и регулируют скорость отбора. К патрубкам через резиновые шланги присоединяют реометры и далее поглотительные приборы.
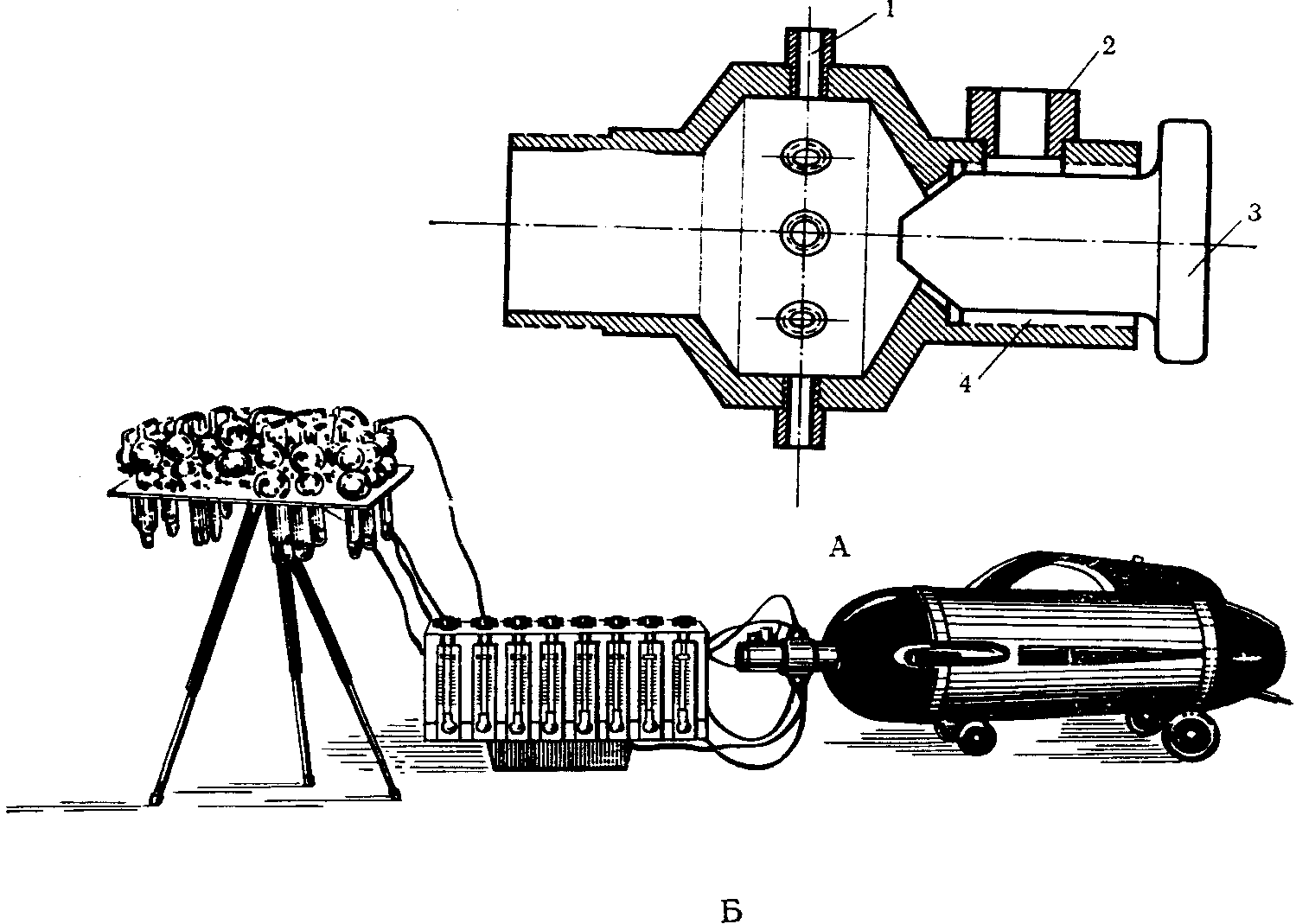
Рис. 13. Установка для отбора проб
А — приставка к пылесосу; Б — общий вид
Для отбора проб атмосферного воздуха довольно широко применяют универсальный электроаспиратор Мигунова-Кабанова (УАМК-3), а также стационарные электроаспираторы — аэрозольный ингалятор АИ-1, портативный аспиратор с аккумуляторными батареями Института общей и коммунальной гигиены имени А.Н. Сысина АМН СССР. Используются также автоматические и неавтоматические электроаспираторы, работающие на переменном токе и аккумуляторах, предложенные Л.Ф. Качором (ЛК-1, ЛК-2, ЛК-3).
ЛК-1 — переносной прибор, может быть установлен на любой автомашине, работает от аккумулятора напряжением 12 В, мощностью 55 Вт. Прибор снабжен ротаметрами, штуцерами для присоединения поглотительных приборов, рассчитан на непрерывное действие в течение 40 мин., после чего выключается для охлаждения. ЛК-2 и ЛК-3 — автоматические стационарные приборы, работающие на переменном токе. ЛК-2 предназначен для отбора разовых проб, снабжен несколькими ротаметрами, работает по заданной программе, позволяет отбирать пробы с интервалами. Прибор может работать и неавтоматически. ЛК-3 позволяет отбирать среднесуточные пробы, работает автоматически по заданной программе. Одновременно можно отбирать три пробы по трем каналам. В сутки по одному каналу проходят до 24 проб, по трем — до 72 [17, 18].
На рис. 14 представлен электрический аспиратор Качора.
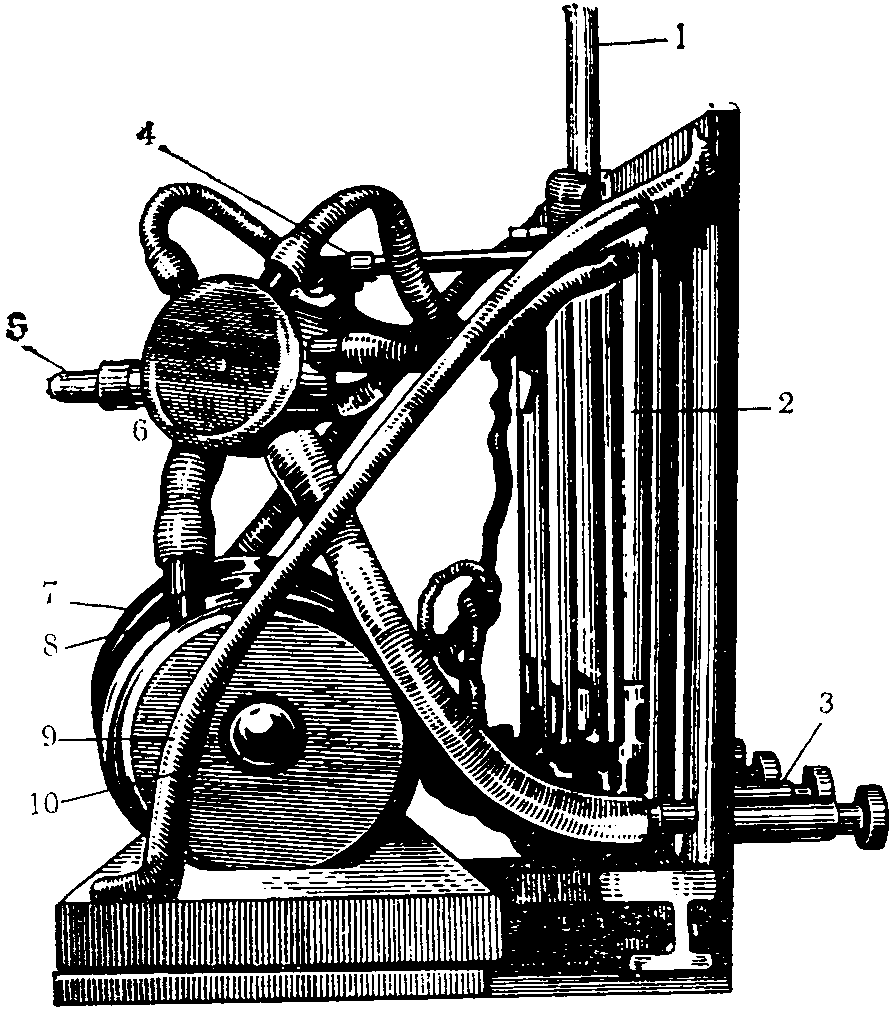
Рис. 14. Электроаспиратор Качора
1 — штатив для поглотительных приборов; 2 — ротаметры;
3 — регуляторы скоростей; 4 — место освещения шкал
ротаметров; 5 — шланги; 6 — воздухосборник;
7 — электромотор; 8, 10 — места смазки;
9 — воздуходувка
Аспирационный отбор проб атмосферного воздуха проводят в различные сезоны года, при повышенных и пониженных температурах. Он связан с протягиванием значительных объемов воздуха. Если поглотительной средой служит жидкость, то при отборе проб при повышенных температурах замечено значительное испарение растворителя, а следовательно, и уменьшение объема поглотительного раствора. Испарение растворителей значительно устраняется применением охладительных смесей, в которые помещают поглотительные приборы.
При отборе проб в условиях пониженных температур часто поглотительные жидкости замерзают. Отбор проб при отрицательных температурах можно проводить, обогревая поглотительные приборы электролампой или простейшим обогревателем, позволяющим в отсутствие электроэнергии поддерживать необходимую температуру.
Обогреватель (рис. 15) представляет собой пластмассовую коробку (а), снабженную двойными стенками. Высота его 180 мм, длина 105 мм, ширина 85 мм. В пространстве между стенками помещен теплоизолирующий слой (б). В верхней части имеется отверстие (в), которое закрывается резиновой пластинкой (г). В резиновой пластинке высверлены отверстие (д) и разрез, куда вставляют поглотительный прибор (е) [12].
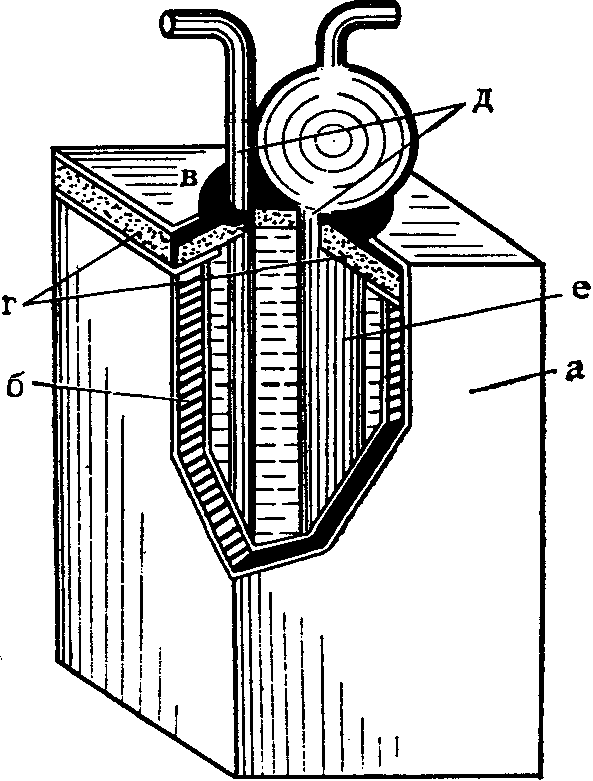
Рис. 15. Обогреватель поглотительных приборов
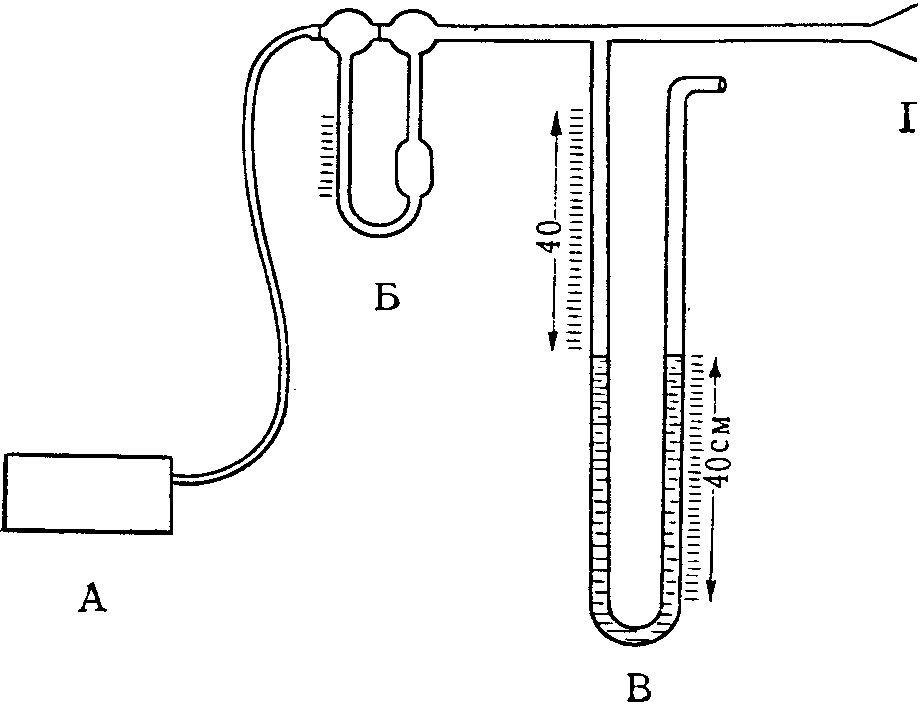
Рис. 16. Схема присоединения поглотительных приборов,
фильтров при измерении сопротивления
А — электроаспиратор; Б — реометр; В — манометр,
Г — место выступления воздуха
Перед отбором обогреватель заполняют водой, нагретой до 20 — 30 °C. При отборе проб со скоростью 1 — 2 л/мин. температура поглотительной жидкости поддерживается до 18 — 20° в течение 2 ч, если температура наружного воздуха находится в пределах 20 °C.
При отборе проб аспирационным методом обычно учитывают сопротивление поглотительных приборов, наполненных раствором или твердым сорбентом, также и фильтров, помещенных в патрон. Измерение сопротивления проводят с помощью водяного манометра, по схеме, указанной на рис. 16.
Отбор проб атмосферного воздуха
в сосуды ограниченной емкости
Отбор проб в ограниченные емкости обычно проводят в тех случаях, когда необходимо установить количество веществ, загрязняющих атмосферный воздух, за короткий промежуток времени. Воздух отбирают в сосуд, объем которого известен. Анализ проводят в малом объеме воздуха, в связи с чем могут быть применены лишь высокочувствительные методы.
Для отбора проб воздуха используют газовые пипетки емкостью до 1 л, а также бутыли различной емкости с пробками и трубками — одной длинной, доходящей до дна, другой короткой (рис. 17, А). Отбор проб может быть осуществлен различными способами: выливанием жидкости, обменным способом, способом вакуумирования воздуха. При использовании способа выливания жидкости емкость предварительно заполняют соответствующим раствором, не реагирующим с исследуемым веществом. В том месте, где берут пробу, жидкость выливают и емкость заполняют исследуемым воздухом. Регулируя скорость вытекания жидкости, удлиняют или укорачивают время отбора пробы.
Применяя обменный способ отбора, пипетку или бутыль присоединяют к аспиратору и на месте отбора пробы емкость продувают десятикратным объемом исследуемого воздуха. Во избежание конденсации паров исследуемого вещества на стенках сосуда продувание ведут со скоростью не менее 2 л/мин.
При отборе проб вакуумным методом применяют сосуды из толстостенного стекла, защищая их чехлом из металлической сетки (рис. 17, 1). Газовую пипетку или бутыль присоединяют к тройнику вакуум-манометра со стороны закрытого колена. Второй конец тройника присоединяют к насосу В (3). Откачивают воздух из емкости до остаточного давления 10 — 15 мм рт. ст. Остаточное давление можно измерить и при помощи открытого манометра, при этом во время откачивания воздуха из емкости один конец ее присоединяют к вакуум-насосу, а второй — к вакуум-манометру (рис. 17, 2). При откачивании воздуха из бутыли ее присоединяют к одной стороне манометра при помощи тройника, а второй конец тройника — к вакуум-насосу. Отбор проб осуществляется путем открывания сосуда в месте исследования атмосферного воздуха.
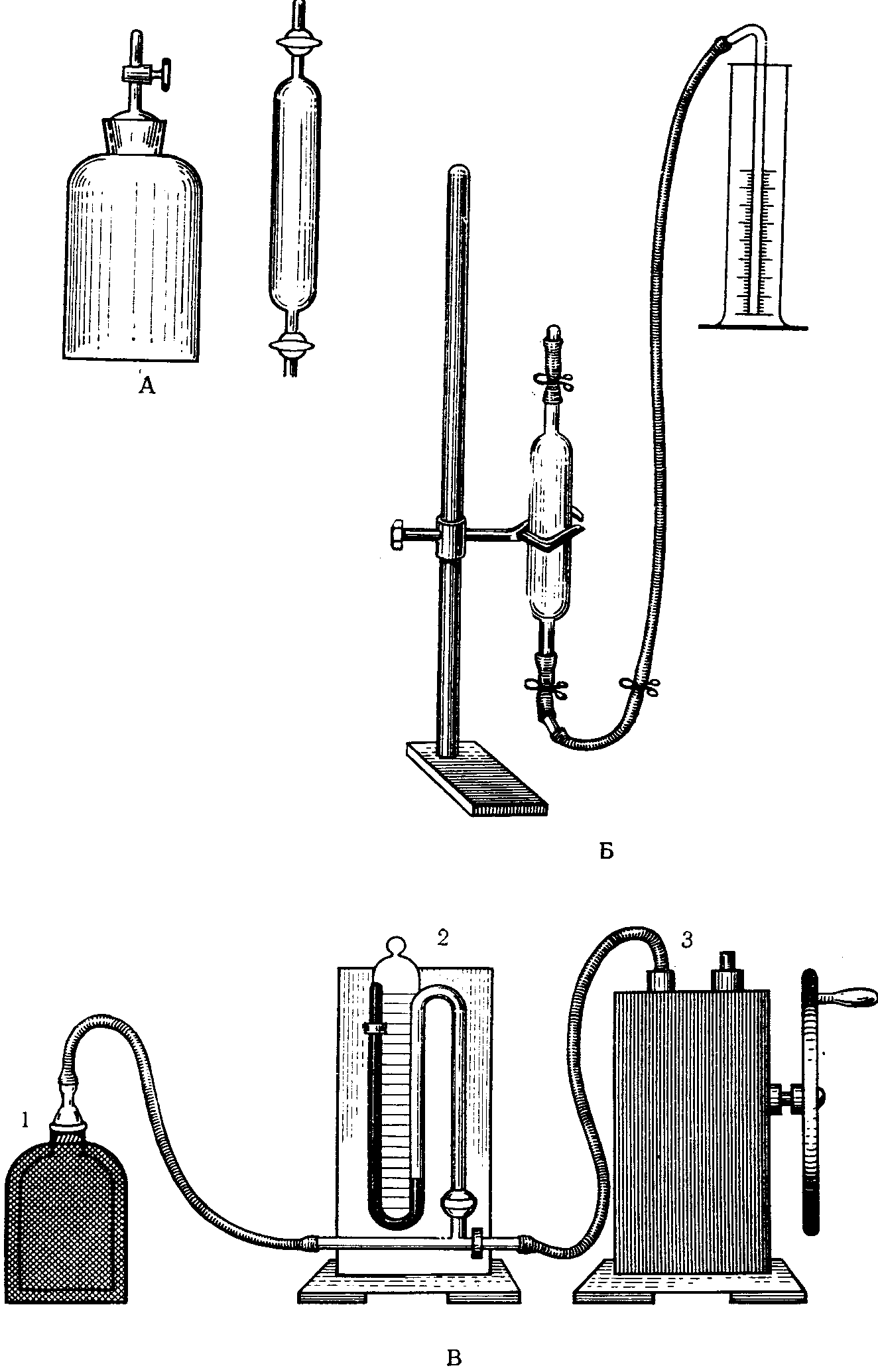
Рис. 17. Приспособления для отбора проб
в сосуды ограниченной емкости
А — бутыль и газовая пипетка; Б — заполнение пипетки
жидкостью; В — схема вакуумирования воздуха из емкости
ИЗМЕРЕНИЕ ОБЪЕМА ИССЛЕДУЕМОГО ВОЗДУХА
Для учета объема и скорости аспирируемого воздуха применяют реометры и другие приборы.
Реометр состоит из стеклянной V-образной манометрической трубки (рис. 18), части которой соединены между собой горизонтальной трубкой, в которую впаяна диафрагма. Манометрической жидкостью является очищенный керосин (удельный вес 0,82) или вода. При пропускании воздуха в той части манометрической трубки, которая находится до диафрагмы, создается давление, а в трубке, находящейся после диафрагмы, — разрежение, способствующее поднятию уровня манометрической жидкости: этот уровень и соответствует объему проходящего воздуха.
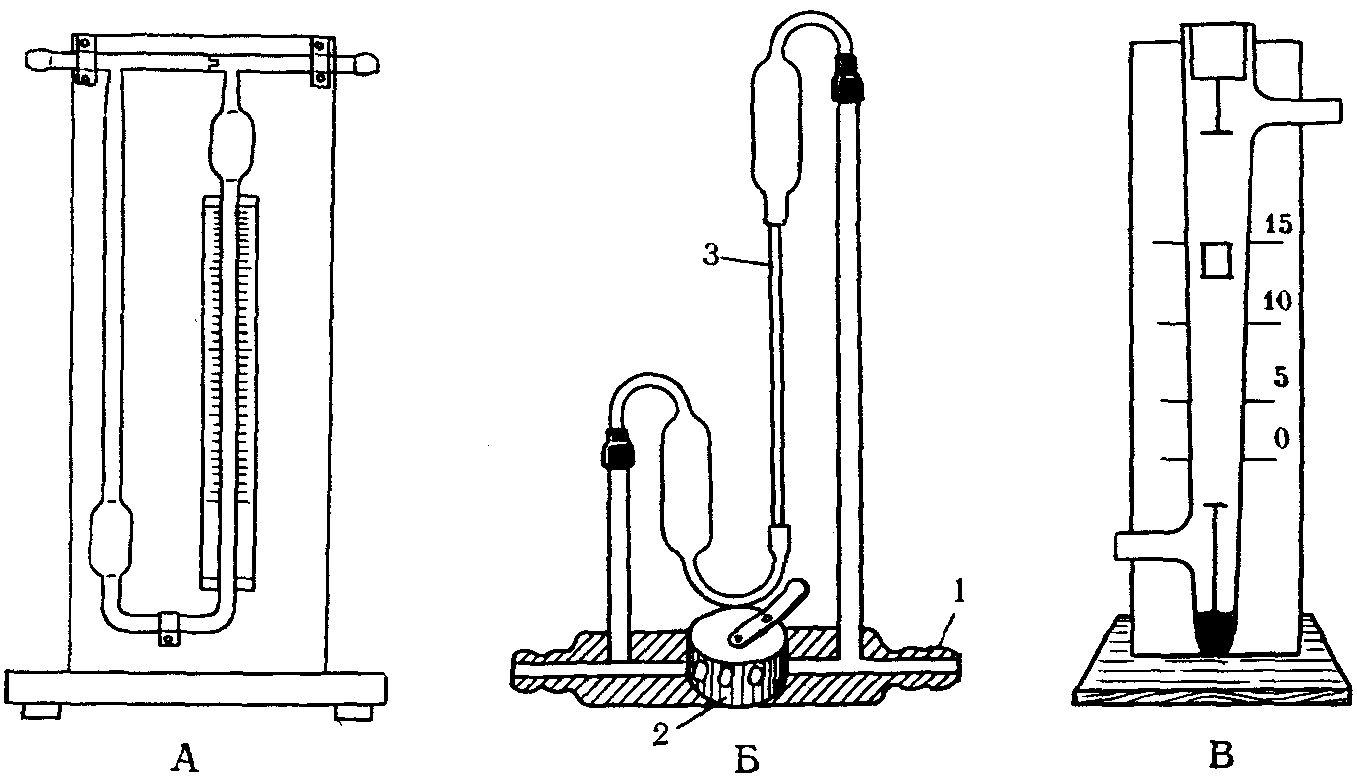
Рис. 18. Приборы, служащие для измерения
скорости прохождения исследуемого воздуха
А — реометр; Б — реометр с поворотными
диафрагмами (1а — место входа воздуха; 2 — шкала;
3 — диафрагма); В — пневмометр
Применяют и реометры с поворотными диафрагмами. Меняя диафрагмы, можно проводить измерение объема воздуха от 0,6 до 80 л/мин. Манометрической жидкостью служит вода. К реометру приложены калибровочные графики, по которым определяют скорость протянутого воздуха в единицу времени.
Могут быть использованы также и сухие ротаметры, представляющие собой вертикально расположенную стеклянную трубку в виде усеченного конуса. Внизу трубки на специальном стержне помещен поплавок строго определенного веса. Поток воздуха поднимает поплавок на уровень, соответствующий скорости движения воздуха (рис. 18). Объем воздуха, взятого для анализа, равен произведению скорости протягивания воздуха в минуту на время протягивания.
При отборе проб отмечают температуру воздуха и атмосферное давление в миллиметрах ртутного столба. В зависимости от сезона года эти данные неидентичны. Для получения сопоставимых результатов объем исследуемого воздуха приводят к нормальным условиям, выражая результат при 0° и давлении 760 мм рт. ст. по формуле:
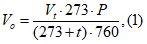
где Vt — объем взятого воздуха при температуре в точке отбора пробы;
Р — атмосферное давление в миллиметрах ртутного столба; t — температура воздуха в местах отбора проб.
Приведенной формулой можно пользоваться при расчете объема воздуха, отобранного аспирационным, обменным способами и способом выливания жидкости.
При вакуумном отборе проб регистрируется остаточное давление в емкости вакуум-манометром или открытым манометром. При использовании вакуум-манометра объем воздуха приводят к нормальным условиям по формуле:
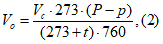
где Vс — объем сосуда; Р — атмосферное давление в миллиметрах ртутного столба; р — остаточное давление в сосуде, отмеченное вакуум-манометром, в миллиметрах ртутного столба, t — температура воздуха в месте отбора проб.
При использовании открытого манометра объем воздуха приводят к нормальным условиям по формуле:
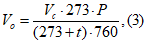
где Vс — объем сосуда; t — температура воздуха у места отбора проб; Р — показание открытого манометра.
Атмосферное давление измеряют в миллиметрах ртутного столба и миллибарах. Бар — давление, равное миллиону дин; 1 мб соответствует 1/1000 бар; 1000 мб соответствует 750, 38 мм рт. ст. Коэффициент пересчета с 1 мм рт. ст. на 1 мб равен 1,33.
Расчет объема воздуха, необходимого для анализа [5, 6]
Концентрации вредных веществ в атмосферном воздухе выражают в миллиграммах на 1 м3 воздуха. Минимальная концентрация токсического вещества, которая может быть определена, зависит от чувствительности применяемого метода. При одновременном присутствии токсических веществ, характеризующихся суммацией действия, когда сумма их концентраций не должна превышать единицы, необходимо определять концентрации веществ ниже предельно допустимых норм, что связано с протягиванием более значительных объемов исследуемого воздуха. Расчет требуемого объема воздуха для анализа проводят по формуле:
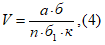
где а — чувствительность определения в микрограммах; б — общий объем пробы в миллилитрах; б1 — объем пробы, взятой для анализа, в миллилитрах; n — предельно допустимая концентрация в миллиграммах на 1 м3 воздуха; к — заданная доля предельно допустимой концентрации.
Расчет анализа проводят по формуле:
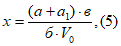
где х — концентрация искомого вещества в миллиграммах на 1 м3 воздуха; а — количество искомого вещества в одном поглотительном приборе, патроне, в микрограммах; а1 — количество искомого вещества во втором поглотительном приборе, патроне, в микрограммах; б — объем пробы, взятой для анализа, в миллилитрах; в — объем всей пробы в миллилитрах; V0 — объем протянутого воздуха, приведенного к нормальным условиям, в литрах.
ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ РАСТВОРОВ
Стандартным раствором называется раствор, содержащий определенное количество вещества в единице объема. Обычно готовят стандартный раствор из вещества, которое определяют в воздухе, или готовят из соединения, в состав которого входит ион искомого вещества. Первоначально готовят исходный стандартный раствор с содержанием вещества порядка десятых миллиграмма в 1 мл. Перед употреблением готовят рабочие стандартные растворы с содержанием сотых, тысячных долей миллиграмма в 1 мл.
Растворителем для приготовления стандартных растворов, как правило, служит поглотительная жидкость, применяемая для поглощения искомого вещества из воздуха.
Готовят стандартные растворы из жидких и твердых веществ в мерных колбах емкостью 100 мл и менее.
Для приготовления стандартного раствора из жидких веществ жидкость предварительно перегоняют и отбирают фракцию погона, отвечающую точке кипения вещества. Первые и последние погоны не используют для приготовления стандартных растворов. Для получения исходного стандартного раствора в мерную колбу вносят 2 — 5 мл растворителя, закрывают пробкой и взвешивают на аналитических весах.
Затем добавляют несколько капель исследуемого вещества и снова взвешивают. По разности весов определяют навеску вещества. Объем доводят до метки, перемешивают содержимое и рассчитывают содержание вещества в 1 мл раствора. Рабочие стандартные растворы готовят соответствующим разбавлением исходных. Вещество для приготовления стандартных растворов из твердых веществ предварительно очищают, перекристаллизовывают или отгоняют (например, фенол). Берут точную навеску и растворяют в мерной колбе.
Если готовят стандартный раствор из соли, то учитывают содержание кристаллизационной воды.
СТАТИСТИЧЕСКАЯ ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗА
При количественном определении токсических веществ в воздухе с целью повышения точности, получения объективной оценки и улучшения качества анализа среди других приемов следует использовать и методы математической статистики. В условиях проведения анализа и при разработке методов определения малых количеств токсических веществ в воздухе приходится ограничиваться малым числом наблюдений. Поэтому для подтверждения статистической достоверности полученных данных используют формулы, пригодные для «малой выборки».
Обычно при анализе выполняют не менее трех — пяти параллельных определений и данные оцениваются как случайная выборка из генеральной совокупности. При обработке результатов наблюдений можно пользоваться следующей схемой. В качестве примера взято определение сернистого газа в воздухе. При анализе получены данные — 10; 12; 10; 12 и 11 мкг. Приведенные величины, будучи расположены в нарастающем порядке, т.е. 10; 10; 11; 12; 12, составляют вариационный ряд, в котором каждое число является вариантой (V) этого ряда.
Находим среднюю арифметическую (М) данного ряда:
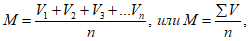
где V1, V2, V3 … до Vn есть значение вариант данного ряда; n — число наблюдений, т.е. число вариант; ![]() — знак суммирования.
— знак суммирования.
Таким образом, в нашем примере:
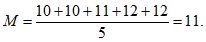
Следующим важным параметром вариационного ряда является среднее квадратичное отклонение (дельта), характеризующее степень различия между отдельными вариантами ряда:
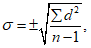
где d — разность между каждой вариантой ряда и полученной средней арифметической величиной, т.е. d = Vn — М.
В нашем примере:
V1 — М = 10 — 11 = -1
V2 — M = 10 — 11 = -1
V3 — M = 10 — 10 = 0
V4 — М = 12 — 11 = +1
V5 — M = 12 — 11 = +1
Как видно, отдельные величины отклоняются от среднего значения, причем они могут быть как положительными, так и отрицательными.
При правильном расчете сумма единичных отклонений должна быть равна 0, т.е. 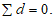 В нашем примере:
В нашем примере:
(-1 — 1 + 1 + 1) = 0.
Далее возводим каждое отклонение в квадрат и находим сумму квадратов отдельных отклонений:
|
d |
d2 |
|
-1 |
1 |
|
-1 |
1 |
|
0 |
0 |
|
+1 |
1 |
|
+1 |
1 |
|
|
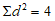
Таким образом:
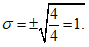
Хотя после извлечения корня квадратного получаются положительные и отрицательные величины, берут только положительное значение.
Для определения статистической достоверности средней арифметической величины используется средняя ее ошибка (mм):
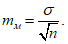
В нашем случае:
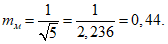
При числе наблюдений менее 30, т.е. когда n < 30, доверительные границы средней арифметической величины, по которым исследователь в каждом случае судит о достоверности полученной средней (М), пользуются выражением:
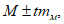
где величина множителя (t), называемая коэффициентом Стьюдента, определяется по таблице. Она показывает, во сколько раз надо увеличить среднюю ошибку, чтобы получить доверительные границы средней арифметической величины с желаемой степенью вероятности (95 или 99,9%).
В приведенном примере при вероятности в 95% t = 2,776.
Таким образом, доверительные границы полученной нами средней арифметической величины:
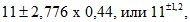
т.е. истинное значение М находится в пределах от 9,8 до 12,2.
Если исследователь считает установленные пределы колебаний средней величины допустимыми, то полученная величина М может считаться достоверной. В противном случае необходимо увеличить число и наблюдений.
Интересным показателем, с точки зрения аналитика, является величина коэффициента вариации ![]() Коэффициент вариации есть отношение среднего квадратичного отклонения
Коэффициент вариации есть отношение среднего квадратичного отклонения ![]() к средней величине (М), выраженное в процентах:
к средней величине (М), выраженное в процентах:
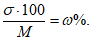
В данном примере коэффициент вариации равен 9,8%. 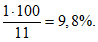
ФОРМА ЗАПИСИ РЕЗУЛЬТАТОВ
|
Наименование статистической характеристики |
Результаты серии опытов |
|
Число определений (n) |
5 |
|
Средняя величина (М) |
11 |
|
Стандартное отклонение отдельных результатов |
1,2 |
|
Средняя ошибка средней величины (tmм) |
0,44 |
|
Коэффициент вариации |
9,8 |
ЛИТЕРАТУРА
1. Алексеева М.В. Методы анализа. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеев Р.И., Коровин Ю.И. Вычисления, производимые при выполнении анализа. В кн.: Руководство по вычислению и обработке результатов количественного анализа. М., «Атомиздат», 1972.
3. Агаджанян И.А. Человек, атмосфера и солнце. М., «Знание», 1968.
4. Атласов А.Г. Определение оптимального объема воздуха при отборе проб воздуха на вредные вещества. Гиг. и сан., 1965, 7, 2.
5. Атласов А.Г. Справочник по отбору проб воздуха на токсические примеси. Проектный институт «Проектпромвентиляция». Ч. 1. М., 1964.
6. Быховская М.С., Гинзбург С.Л., Хализова О.Д. Отбор проб воздуха. В кн.: Определение вредных веществ в воздухе. М., «Медицина», 1966.
7. Бабенко А.С., Володченко Т.Т. Применение цилиндрических кювет для снижения определяемого минимума при измерениях на ФЭК-М. Известия высших учебных заведений. Химия и химическая технология, 1968, в. 10, т. II.
8. Басманов П.И., Борисов Н.Б. В Бр. Фильтры АФА. Каталог-справочник. М., «Атомиздат», 1970.
9. Бланк А.Б. Простое приспособление для снижения определения при измерении на фотоэлектроколориметре ФЭК-М, ФЭК-Н. Журнал аналитической химии АН СССР, 1962, т. 17, в. 8.
10. Близнев В.И. Приспособление к санитарной машине УАЗ-450а для отбора атмосферного воздуха. Гиг. и сан., 1964, 5.
11. «В интересах народа». Передовая статья газеты «Правда» от 23 сентября 1972 г.
12. Гильденскиольд Р.С., Рихтер Б.В. Полевой обогреватель для поглотителей при отборе проб воздуха. Гиг. и сан., 1968, 2.
13. Доерфель К. Приемы вычислений. В кн.: Статистика в аналитической химии. М., «Мир», 1969.
14. Инструктивно-методические указания по организации исследования атмосферного воздуха. Медицинская литература. М., 1963.
15. Качмар Е.Г., Хрусталева В.А. Применение твердых зерненных сорбентов при исследовании атмосферного воздуха. Гиг. и сан., 1969, 9.
16. Качмар Е.Г., Хрусталева В.А. Определение вредных веществ в атмосферном воздухе с применением твердых зерненных сорбентов. Московский научно-исследовательский институт гигиены им. Ф.Ф. Эрисмана. М., 1964.
17. Качор Л.Ф. Конструкция аспираторов для отбора проб пыли и газов в атмосферном воздухе. Гиг. и сан., 1950, 4.
18. Качор Л.Ф. Новая аппаратура для отбора проб атмосферного воздуха. Гиг. и сан., 1964, 9.
19. Коренман И.М. Введение. В кн.: Введение в количественный ультрамикроанализ. М., «Госхимиздат», 1968.
20. Коренман И.М. Некоторые особенности очень малых разбавленных растворов. В кн.: Аналитическая химия малых концентраций. М., «Химия», 1968.
21. Ноткин Е.Л. Математическая статистика в гигиенических исследованиях. Вопросы санитарной статистики. Ученые записки N 10 Московского научно-исследовательского института гигиены им. Ф.Ф. Эрисмана. М., 1962.
22. О мерах по дальнейшему улучшению охраны природы и рациональному использованию природных ресурсов. Постановление Верховного Совета СССР. Газета «Правда» N 265 от 21 сентября 1972 г.
23. О мерах по дальнейшему улучшению здравоохранения и развитию медицинской науки в стране. Постановление ЦК КПСС и Совета Министров СССР от 5 июля 1968 г. N 517.
24. Основы законодательства Союза ССР и союзных республик о здравоохранении. «Медицинская газета» от 23 декабря 1969 г.
25. Предельно допустимые концентрации вредных веществ в атмосферном воздухе населенных мест. Санитарные нормы проектирования промышленных предприятий. СН-245-71. М., 1972.
26. Решения XXIV съезда КПСС.
27. Семенов В.А., Попов Е.И. Приставка к пылесосу для отбора проб атмосферного воздуха. Гиг. и сан., 1965, 8.
28. Шпинькова Л.И. Фильтры АФА. М., «Изотоп», 1972.
Глава II
ГАЛОГЕНЫ И ГАЛОИДОВОДОРОДЫ
ФТОРИСТЫЙ ВОДОРОД
Бесцветный газ с резким запахом, хорошо растворимый в воде. Плотность по отношению к воздуху 0,713, температура кипения 19,5°, температура плавления -81°, температура замерзания -179°. Фтористоводородная (плавиковая) кислота, бесцветная жидкость, дымящая во влажном воздухе, смешивающаяся с водой в любых соотношениях, плотность 1,15, содержит 35,3% фтористого водорода; как и фтористый водород, реагирует со многими металлами и кремниевым ангидридом (SiO2), сохраняется в парафинированных сосудах.
Фтористый водород сильно раздражает верхние дыхательные пути, обладает также общей токсичностью.
Предельно допустимые концентрации газообразных фтористых соединений (HF, SiF4) (в пересчете на фтор): максимальная разовая 0,02 мг/ м3, среднесуточная 0,005 мг/ м3.
ОПРЕДЕЛЕНИЕ ФТОРИСТОГО ВОДОРОДА
С ТИТАНХРОМОТРОПОВЫМ РЕАКТИВОМ [6]
Принцип определения
При взаимодействии иона фтора с титанхромотроповым комплексом образуется титанфторидное соединение. Окраска раствора в зависимости от концентрации иона фтора изменяется от темно- до светло-розовой, малодиссоциируемого титанфторидного соединения.
Чувствительность определения 2,0 мкг иона фтора в анализируемом объеме раствора. Определению не мешают сернистый газ, серная кислота.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартные растворы фторида натрия. Исходный стандартный раствор с содержанием в 1 мл 100 мкг иона фтора готовят растворением 0,0221 г перекристаллизованного фторида натрия в 100 мл воды в мерной колбе. Рабочий стандартный раствор с содержанием 10 мкг/мл иона фтора готовят разведением исходного стандартного раствора в 10 раз. Очистка фторида натрия — см. примечание.
3. Двуокись титана.
4. Сульфат титана.
Для получения сульфата титана 20 г пиросернокислого калия и 1 г двуокиси титана сплавляют в кварцевом или фарфоровом тигле емкостью 100 — 200 мл при температуре не выше 800° в течение нескольких часов до получения прозрачного сплава. После остывания сплав заливают 5% раствором серной кислоты и оставляют до следующего дня. Сернокислый титан растворяют при комнатной температуре в 500 мл 5% раствора серной кислоты, все время помешивая, так как растворение идет медленно. После растворения раствор фильтруют через бумажный фильтр, раствор должен быть прозрачным.
Проверяют содержание сульфата титана в 1 мл раствора весовым методом. Для этого в 3 стакана емкостью по 50 мл каждый приливают по 10 мл приготовленного раствора, добавляют по 2 мл 10% раствора хлористого аммония, 1 каплю метилового оранжевого и по каплям 25% раствор аммиака до слабого запаха. Титан выпадает в виде хлопьев гидроокиси титана.
Осадок отфильтровывают через беззольный фильтр и промывают 2% раствором азотнокислого аммония до удаления иона сульфата. Фильтры с осадками озоляют, прокаливают при температуре 1000 — 1100 °C и взвешивают двуокись титана. Определяют содержание двуокиси титана в растворе. Содержание должно быть около 2 мг двуокиси титана или около 6 мг сернокислого титана в 1 мл приготовленного раствора. Раствор годен к употреблению в течение нескольких лет. Его хранят в темном месте.
5. Пиросульфат калия (натрия).
6. Серная кислота, 5% раствор.
7. Аммиак, 25% раствор.
8. Нитрат аммония, 2% раствор.
9. Хромотроповая кислота или ее динатриевая соль. 20 мг кислоты растворяют в 10 мл воды перед использованием.
10. Монохлоруксусная кислота.
11. Едкий натр, 0,1 и 0,005 н. растворы.
12. Буферный раствор с pH 2,85. 0,95 г монохлоруксусной кислоты растворяют в 100 мл воды, к раствору приливают 50 мл 0,1 н. раствора едкого натра.
13. Составной раствор. В мерную колбу емкостью 100 мл вносят 4,8 мг (или 0,8 мл раствора) сернокислого титана, 10 мл раствора хромотроповой кислоты, 20 мл буферной смеси, доводят водой до метки и перемешивают. Составной раствор, окрашенный в вишневый цвет, годен к употреблению в течение месяца.
14. Парафин.
15. Метиловый оранжевый, 0,5% раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 8 — 10 л/мин. протягивают через 2 последовательно соединенные поглотительные прибора Рыхтера, содержащие по 6 мл воды. Внутреннюю поверхность первого поглотительного прибора парафинируют. Продолжительность отбора 20 — 30 мин.
Ход анализа
5 мл пробы из каждого поглотительного прибора переносят в пробирки и проверяют pH среды. Если раствор имеет кислую реакцию, то его нейтрализуют 0,005 н. раствором едкого натра с индикатором метиловым оранжевым. В фарфоровой чашке к 0,005 мл исследуемого раствора добавляют 1 каплю метилового оранжевого и, если раствор окрашивается в розовый цвет, приливают из микробюретки 0,005 н. раствор едкого натра до окраски, равной окраске контроля (вода с одной каплей индикатора). Зная, сколько потребовалось щелочи на 0,5 мл раствора, прибавляют к 5 мл 10-кратное количество щелочи, взбалтывают и берут 5 мл для анализа. Затем к пробам приливают по 3 мл составного раствора, хорошо перемешивают и через 10 мин. фотометрируют в кюветах с толщиной слоя 2 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание иона фтора в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 3).
Таблица 3
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,7 |
1,0 |
1,5 |
2,0 |
|
Вода, мл |
5 |
4,8 |
4,6 |
4,3 |
4,0 |
3,5 |
3,0 |
|
Содержание иона фтора, мкг |
0 |
2 |
4 |
7 |
10 |
15 |
20 |
С увеличением содержания иона фтора окраска растворов ослабевает. Все пробирки шкалы обрабатывают аналогично пробам и измеряют оптическую плотность.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами. В пробирках с притертыми пробками шкала устойчива в течение месяца.
Расчет см. выше.
Примечания. 1. Очистка фторида натрия. В 1 л горячей воды растворяют 45 г фторида натрия, добавляют 15 г хлорида калия и оставляют на ночь. Раствор фильтруют и выпаривают в платиновой чашке на водяной бане примерно до 40 мл жидкости. Кристаллы фторида натрия отсасывают и промывают холодной дистиллированной водой до отрицательной реакции на ион хлора. Фторид натрия сушат при 120 — 150 °C и хранят в стеклянной банке с притертой пробкой.
2. Предложенный метод определения фтористого водорода может быть использован и для определения других неорганических газообразных соединений фтора.
ОПРЕДЕЛЕНИЕ ФТОРИСТОГО ВОДОРОДА С ЭРИОХРОМЦИАНИНОМ [5]
Принцип определения
Ион фтора в щелочной среде вызывает ослабление окраски соединения, образующегося при взаимодействии соединений фтора с эриохромцианином и хлористым цирконием.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Реактивы
1. Поглотительный раствор. 20 мл 0,1 н. раствора едкого натра разбавляют до 1 л дистиллированной водой.
2. Исходный стандартный раствор фтористого натрия.
0,1105 г фтористого натрия растворяют в дистиллированной воде и объем доводят водой до 500 мл. Рабочий стандартный раствор с содержанием 10 мкг иона фтора в 1 мл готовят разбавлением водой исходного раствора.
3. Едкий натр, 0,1 н. раствор.
4. Эриохромцианин-R.
0,108 г эриохромцианина-R растворяют в 100 мл дистиллированной воды.
5. Хлорокись циркония.
В небольшом количестве дистиллированной воды растворяют 125 мг хлорокиси циркония, затем добавляют 15 мл соляной концентрированной кислоты и объем доводят водой до 100 мл.
Отбор пробы
Для определения разовой концентрации исследуемый воздух со скоростью 3 — 5 л/мин. просасывают через поглотительный прибор Рыхтера, наполненный 10 мл поглотительного раствора.
Ход анализа
Содержимое поглотительного прибора доводят дистиллированной водой до первоначального объема. Переносят пробу в пробирку, добавляют 0,5 мл раствора эриохромцианина, 1 мл раствора хлорокиси циркония, взбалтывают и нагревают в течение 10 мин. при 20 °C. Затем окраски растворов фотометрируют при длине волны 530 нм в кювете с толщиной слоя 1 — 2 см по отношению к контролю, который готовят одновременно и аналогично пробе. Содержание иона фтора в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику. Для построения градуировочного графика готовят шкалу стандартов (табл. 4).
Таблица 4
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
1 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
10 |
9,9 |
9,8 |
9,6 |
9,4 |
9,2 |
9,0 |
8,0 |
|
Содержание иона фтора, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Шкалу стандартов обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
ХЛОР
Газ желтовато-зеленого цвета, обладает резким раздражающим запахом, плотность при 0° — 2,49 (по воздуху).
Хлор легко сжижается в желтовато-зеленую жидкость под давлением 6 атм., плотность жидкости 1,55 при 34,1°.
Растворим в воде, хлороформе, четыреххлористом углероде и других органических растворителях. Сорбируется активированным углем.
Хлор действует раздражающе на дыхательные органы и вызывает отек легких.
Предельно допустимые концентрации: максимальная разовая 0,10 мг/м3, среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРА С МЕТИЛОВЫМ ОРАНЖЕВЫМ [2, 3]
Принцип определения
При взаимодействии хлора в кислой среде с метиловым оранжевым ослабляется розовая окраска раствора. Уменьшение интенсивности окраски пропорционально количеству хлора.
Чувствительность определения — 0,2 мкг в анализируемом объеме раствора.
Сернистый газ, окислы азота до 10 мг/м3, а озон в концентрациях до 5 мг/м3 не мешают определению.
Реактивы
1. Поглотительный раствор: готовят исходный раствор. В мерной колбе емкостью 1 л растворяют 0,1 г метилового оранжевого в 100 мл горячей воды, по охлаждении прибавляют 0,5 г бромида калия х.ч., 20 мл этанола, содержание колбы перемешивают и добавляют воды до метки.
Раствор устойчив.
Для приготовления рабочего поглотительного раствора исходный раствор разбавляют. В мерную колбу емкостью 100 мл вливают 4 мл 0,05 н. серной кислоты и 5 мл исходного раствора и доводят до метки водой; 25 мл этого раствора помещают в коническую колбу и титруют рабочим стандартным раствором хлора до лимонно-желтой окраски. На 25 мл рабочего поглотительного раствора должно расходоваться 2,5 — 3 мл рабочего стандартного раствора хлора.
Если на титрование расходуется больше или меньше стандартного раствора хлора, то необходимо поглотительный раствор соответствующим образом разбавить или приготовить более концентрированный раствор.
Хранить его следует в темном месте.
2. Стандартный раствор хлора.
Готовят исходный раствор: 5 г хлорной извести растворяют в 250 мл воды. Раствор оставляют в покое на несколько часов и прозрачный раствор фильтруют в мерную колбу емкостью 1 л, доводят до метки дважды перегнанной водой, переливают в темную склянку и определяют титр раствора титрованием его 0,02 н. раствором тиосульфата.
Перед применением раствора каждый раз определяют содержание хлора йодометрически. Для титрования берут 10 мл исходного раствора хлора в коническую колбу с притертой пробкой емкостью 250 мл, вливают 5 мл 10% серной кислоты, 60 мл воды, всыпают 1 г йодида калия, раствор взбалтывают и оставляют в темном месте на 5 — 10 мин. Затем титруют свежеприготовленным 0,02 н. раствором тиосульфата до образования бледно-желтой окраски, вливают в раствор 1 мл 0,5% крахмала и титруют до обесцвечивания. Содержание хлора определяют из расчета: 1 мл 0,02 н. раствора тиосульфата соответствует 0,709 мг хлора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора. В мерную колбу емкостью 100 мл вливают такое количество исходного раствора, в котором содержалось бы 1000 мкг хлора, и доводят до метки дважды перегнанной водой. Перед применением проверяют содержание хлора йодометрически, титруют раствор 0,002 н. раствором тиосульфата, который готовят перед титрованием. 1 мл 0,002 н. раствора тиосульфата соответствует 0,0709 мг хлора.
3. Метилоранжевый, х.ч.
4. Калий бромид, х.ч.
5. Этанол 96°, х.ч.
6. Серная кислота, 0,05 н. раствор.
7. Хлорамин Б.
8. Йод возогнанный, растертый и высушенный.
9. Тиосульфат, 0,1 н. раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащим 2,5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. При изменении окраски поглотительного раствора отбор прекращают.
б) Для определения среднесуточной концентрации воздух со скоростью 5 л/ч протягивают в течение суток через поглотительный прибор с пористым фильтром N 1, содержащий 5 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Из поглотительного прибора жидкость выливают в колориметрическую пробирку. Одновременно строят шкалу стандартов (табл. 5). Пробу колориметрируют на белом фоне.
Таблица 5
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,10 |
0,15 |
0,20 |
0,25 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,95 |
1,90 |
1,85 |
1,80 |
1,75 |
|
Содержание хлора, мкг |
0 |
0,20 |
0,50 |
1,0 |
1,5 |
2,0 |
2,5 |
Шкала сохраняется в течение месяца в темном месте.
Расчет см. выше.
Примечание. Можно концентрацию хлора установить фотометрически. Для этого готовят шкалу стандартов с содержанием 1, 2, 3 и 4 мкг хлора в объеме 5 мл. Измеряют величины оптических плотностей полученных растворов и строят калибровочный график. В качестве контроля используют поглотительный раствор, но предварительно измеряют его оптическую плотность по отношению к дистиллированной воде. Перед отбором проб проверяют оптическую плотность поглотительного раствора. Если величина оптической плотности изменилась, то раствор разбавляют водой или добавляют в него исходного поглотительного раствора до получения первоначальных данных.
ХЛОРИСТЫЙ ВОДОРОД И СОЛЯНАЯ КИСЛОТА
Хлористый водород — бесцветный газ с резким запахом, плотность по отношению к воздуху 1,26, вес 1 л его при 0 °C 1,6391.
Хлористый водород поглощается водой, один объем воды при комнатной температуре и атмосферном давлении может растворить 450 объемов хлористого водорода. Он растворяется в эфире и спирте. Во влажном воздухе хлористый водород образует аэрозоль соляной кислоты. Хлористый водород и аэрозоль соляной кислоты раздражают верхние дыхательные пути, вызывают воспаление слизистой оболочки глаз и помутнение роговицы.
Предельно допустимые концентрации: максимально разовая по молекуле — 0,2 мг/м3, по иону водорода — 0,006 мг/м3, среднесуточная по молекуле — 0,2 мг/м3, по иону водорода — 0,006 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРИСТОГО ВОДОРОДА И СОЛЯНОЙ КИСЛОТЫ
С НИТРАТОМ СЕРЕБРА [1]
Принцип определения
При взаимодействии иона хлора с нитратом серебра образуется взвешенная муть. По интенсивности помутнения определяют содержание иона хлора.
Чувствительность определения — 2 мкг в анализируемом объеме. Соли других галоидов и синильной кислоты мешают определению.
Реактивы
1. Поглотительный раствор — дважды перегнанная вода.
2. Стандартный раствор хлористого водорода. Исходный раствор с содержанием 100 мкг/мл готовят растворением 0,0204 г хлорида калия в воде в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз.
3. Нитрат серебра, 1% раствор (хранят в темной склянке).
4. Азотная кислота, 10% раствор.
Все реактивы готовят на дважды перегнанной воде, свободной от иона хлора.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 1 л/мин. через два поглотительных прибора с пористым фильтром N 1, содержащие по 6 мл дважды перегнанной воды. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 10 л/ч протягивают через вышеуказанный поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, периодически добавляют водой до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В пробирку переносят 2,5 — 3 мл пробы, вливают 1 мл 10% азотной кислоты и 0,5 мл нитрата серебра. Содержимое пробы встряхивают, через 10 мин. помещают в кювету шириной 1 см и сравнивают с контролем с помощью фотоэлектроколориметра при длине волны 364 нм.
Содержание вещества в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 6).
Таблица 6
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
2,5 |
2,3 |
2,1 |
1,9 |
1,7 |
1,5 |
0,5 |
|
Содержание иона хлора, мкг |
0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
20,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СОЛЯНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА [2, 4]
Принцип определения
Метод основан на измерении оптической плотности при длине волны 530 нм окрашенного водного раствора нацело диссоциированной кислоты с метиловым красным в интервале значения pH 4,7 — 5,3.
Чувствительность определения — 0,05 мкг ионов водорода в анализируемом объеме (5 мл).
Определению мешают основания и кислоты, реагирующие с метиловым красным в указанном интервале pH.
Присутствие CO2 в количестве 0,05 об.% и SO2 в количестве 0,5 мг/м3 не мешает определению.
Аппаратура
1. Спектрофотометр, позволяющий проводить измерение оптической плотности в видимой области спектра, или фотоэлектроколориметр.
2. Поглотительные приборы Зайцева.
Реактивы
1. Дистиллированная вода, ГОСТ 6709-53.
Ее проверяют добавлением к 5 мл воды 1 капли метилового красного и 1 капли 0,005 н. раствора соляной кислоты. При pH 5,8 — 6,6 раствор окрашивается в розовый цвет.
2. 0,1 н. раствор соляной кислоты. Перед употреблением готовят исходный 0,005 н. раствор соляной кислоты с содержанием 5 мкг/мл ионов водорода.
Рабочий стандартный раствор с содержанием 0,5 мкг ионов водорода в 1 мл готовят разбавлением исходного в 10 раз.
3. 0,01% раствор метилового красного.
Отбор пробы
Для определения разовой концентрации исследуемый воздух протягивают через поглотительный прибор, наполненный 6 — 10 мл дистиллированной воды, со скоростью 0,5 л/мин. в течение 20 мин.
По окончании отбора объем пробы доводят до первоначального.
Ход анализа
К 5 мл испытуемого раствора добавляют 0,2 мл раствора метилового красного, помещают в кювету шириной 1 — 2 см и через 1 — 1 1/2 мин. замеряют оптическую плотность по сравнению с контролем при длине волны 530 нм.
Содержание иона водорода в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика в пробирках готовят шкалу стандартов (табл. 7).
Таблица 7
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,15 |
0,2 |
0,3 |
0,4 |
|
Раствор метилового красного, мл |
По 0,4 мл |
|||||
|
Дистиллированная вода, мл |
10 |
9,5 |
9,45 |
9,4 |
9,3 |
9,2 |
|
Содержание ионов водорода, мкг |
0 |
0,05 |
0,07 |
0,1 |
0,15 |
0,2 |
Содержимое пробирок перемешивают, измеряют оптическую плотность и строят график.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение хлористого водорода и соляной кислоты. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Вильнер Т.А. К определению хлора с метиловым оранжевым в атмосферном воздухе. Гиг. и сан., 1972, 6.
3. Криворучко Ф.Д. Определение хлора в атмосферном воздухе. Гиг. и сан., 1953, 3.
4. Манита М.Д. Спектрофотометрический метод определения азотной и соляной кислот в присутствии нитратов и хлоридов в атмосферном воздухе. Гиг. и сан., 1964, 3.
5. Определение фтористого водорода с эриохромцианином. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
6. Панин К.П. Определение фтористого водорода (Фотометрический метод). Гиг. и сан., 1967, 12.
Глава III
СОЕДИНЕНИЯ СЕРЫ
СЕРНИСТЫЙ АНГИДРИД (СЕРНИСТЫЙ ГАЗ)
Бесцветный газ, с резким характерным запахом, который не поддерживает горение и сам не горит.
Плотность при нормальных условиях по отношению к воздуху 2,2630.
Вес 1 л газообразного SO2 2,9256 г.
Сернистый газ хорошо растворим в воде: в 100 г воды при 20 °C растворяется 10,5 г, при 10 °C — 15,4 г, при 0 °C в одном объеме воды — 70 объемов сернистого газа. Водный раствор имеет кислую реакцию.
Сернистый газ обладает восстановительными свойствами.
Действуя на организм, сернистый газ раздражает дыхательные пути, слизистые оболочки глаз, вызывает кашель.
Предельно допустимые концентрации: максимальная разовая доза 0,5 мг/м3; среднесуточная 0,15 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА
С ФУКСИНФОРМАЛЬДЕГИДНЫМ РЕАКТИВОМ [1, 2]
Принцип определения
Сернистый ангидрид улавливают в раствор тетрахлормеркурата натрия с прибавлением комплексона (для предотвращения каталитического окисления). Образующееся соединение дает в кислой среде с фуксином и формальдегидом красно-фиолетовую окраску, интенсивность которой пропорциональна количеству сернистого газа.
Чувствительность определения — 0,3 мкг в анализируемом объеме раствора.
Определению не мешают хлор, озон в концентрациях, превышающих содержание сернистого ангидрида. Сероводород устраняют в виде сульфида ртути, отделяя его фильтрованием или центрифугированием.
Двуокись азота мешает определению.
Реактивы
1. Поглотительный раствор; 0,1 М раствор тетрахлормеркуроата натрия и 0,01% раствор комплексона III (динатриевой соли этилендиаминотетрауксусной кислоты) готовят путем растворения 27,2 г HgCl2, 11,7 г NaCl и 0,1 г комплексона III в 1 л дистиллированной воды.
2. Исходный стандартный раствор сульфита. 0,1968 г безводного сульфита натрия х.ч. или 0,1735 г пиросульфита калия (K2S2O5) растворяют в 1 л поглотительного раствора. В 1 мл раствора содержится 100 мкг сернистого газа.
3. Рабочий стандартный раствор. Разбавляют 5 мл исходного раствора до объема 100 мл поглотительным раствором. 1 мл такого раствора содержит 5 мкг сернистого газа.
4. Раствор фуксина: 0,65 г основного фуксина (кристаллического) х.ч. растворяют после размельчения в 22 мл этанола, разбавляют 100 мл дистиллированной воды, медленно и после охлаждения добавляют 60 мл концентрированной серной кислоты х.ч. и после охлаждения осторожно доводят водой до метки 1 л.
5. 2% раствор формальдегида х.ч. Для этого 5 мл 40% раствора формальдегида доводят водой до 100 мл.
6. Фуксинформальдегидный реактив: 10 частей свежепрофильтрованного раствора фуксина смешивают с 1 частью раствора формальдегида. Реактив применяют свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 10 л/ч протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же прибор. Пробы должны быть проанализированы в течение 48 ч.
Ход анализа
В пробирку переносят 3 мл пробы, добавляют 2 мл фуксинформальдегидного реактива, содержимое пробирки перемешивают, через 20 мин. переносят в кювету шириной 1 см и измеряют оптическую плотность по сравнению с контролем с помощью фотоэлектроколориметра при длине волны 575 нм. Содержание вещества в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 8).
Таблица 8
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,06 |
0,12 |
0,18 |
0,24 |
0,30 |
0,6 |
0,9 |
|
Поглотительный раствор, мл |
3 |
2,94 |
2,88 |
2,82 |
2,76 |
2,7 |
2,4 |
2,1 |
|
Содержание сернистого газа, мкг |
0 |
0,3 |
0,6 |
0,9 |
1,2 |
1,5 |
3,0 |
4,5 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график. Для визуального определения строят шкалу стандартов (табл. 9).
Таблица 9
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,06 |
0,1 |
0,2 |
0,4 |
0,8 |
1,2 |
1,6 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,94 |
1,9 |
1,8 |
1,6 |
1,2 |
0,8 |
0,4 |
— |
|
Содержание сернистого газа, мкг |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10 |
Для анализа берут 2 мл пробы.
Во все пробирки вливают по 1 мл фуксинформальдегидного реактива, растворы перемешивают. Определение проводят через 20 мин. после вливания реактива. Шкалу и пробу обрабатывают одновременно.
Расчет см. выше.
Примечания: а) При наличии окислов азота применяют поглотительный раствор, содержащий 0,06% аминосульфоновой кислоты или 0,08% аминосульфоната калия (NH2SO3K).
б) Раствор фуксина не должен иметь в диапазоне 600 — 750 нм в кювете шириной 1 см по сравнению с дистиллированной водой большую оптическую плотность, чем 0,05. При более высоких величинах оптической плотности раствор фуксина следует очистить. Раствор встряхивают 2 раза с бензолом, затем прибавляют на 1 л раствора до 20 г активированного угля, тщательно встряхивают в течение 1/2 — 1 ч и фильтруют через плотный аналитический фильтр («синяя лента»).
В случае необходимости раствор фильтруют через фильтр с активированным углем несколько раз до тех пор, пока оптическая плотность не достигнет нужных величин. Правильно приготовленный раствор окрашен в оранжево-желтый цвет, его оптическая плотность по сравнению с дистиллированной водой в кювете 1 см должна быть 0,02 — 0,03.
в) Для точной навески сульфита или пиросульфита при приготовлении исходного раствора следует установить точное содержание SO2 в применяемой соли путем йодометрического титрования.
г) В качестве поглотительного раствора можно применять 0,01 н. раствор едкого натра в 5% растворе глицерина.
Если же в качестве поглотительного раствора был взят 0,01 н. раствор едкого натра в 5% растворе глицерина, то сероводород, присутствующий в воздухе, удаляют с помощью твердого зернистого сорбента (зерна 4 — 5 мм) 10 мл пемзы, пропитанной насыщенным раствором ацетата свинца или сульфатом меди, высушенной при 105 — 110° и помещенной в стеклянную трубку. Последнюю присоединяют к входному отверстию поглотительного прибора в вертикальном положении [3].
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА С ПАРАРОЗАНИЛИНОМ [2]
Принцип определения
При взаимодействии сернистого газа с парарозанилином и формальдегидом в кислой среде раствор окрашивается в красно-фиолетовый цвет, по интенсивности которого определяют содержание сернистого газа.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Двуокись азота мешает определению в концентрациях 4 мг/м3.
Реактивы
1. Поглотительный раствор 0,1 М раствор тетрахлормеркурата натрия: в мерной колбе емкостью 1 л, наполненной 800 мл воды, растворяют 11,7 г хлорида натрия и затем 27,2 г хлорида ртути х.ч. После полного растворения объем раствора в колбе доводят до метки водой.
Через несколько часов раствор фильтруют через плотный стеклянный фильтр и прозрачный раствор плотно закрывают; он устойчив в течение длительного времени. Раствор ядовит.
2. Стандартный раствор сернистого газа: исходный раствор с содержанием 4 мг/мл получают растворением 0,5936 г пиросульфита натрия х.ч. водой в мерной колбе емкостью 100 мл. Раствор применяют свежеприготовленный. Предварительно устанавливают содержание сернистого газа в 1 г соли.
Стандартный раствор А с содержанием 20 мкг/мл сернистого газа получают из 0,5 мл исходного раствора, который вливают в мерную колбу емкостью 100 мл и доводят раствор до метки поглотительным раствором. Раствор применяют всегда свежеприготовленным.
Рабочий стандартный раствор с содержанием 2 мкг/мл сернистого газа получают из стандартного раствора А при разбавлении его в 10 раз. Раствор применяют всегда свежеприготовленный.
3. Парарозанилин-хлоргидрат, 0,04% раствор: готовят в мерной колбе емкостью 500 мл, в которую наливают 400 мл воды и вносят 0,200 г парарозанилина-хлоргидрата х.ч. и растворяют. Через сутки прибавляют 30 мл соляной кислоты уд. веса 1,19, раствор доводят до метки водой и тщательно встряхивают. По истечении суток раствор готов. Хранят его в темном месте при нормальной температуре в плотно закрытой склянке. Раствор устойчив в течение месяца.
4. Формальдегид, 0,2% раствор: 2,5 мл 40% раствора формальдегида х.ч. разбавляют водой в мерной колбе емкостью 50 мл. Раствор устойчив в течение 2 нед.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 10 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух непрерывно в течение суток протягивают со скоростью 25 л/ч через поглотительный прибор с пористым фильтром N 1, в котором содержится 10 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же поглотительный прибор в течение 30 мин.
Отобранные пробы можно хранить в течение недели в темноте при комнатной температуре.
Ход анализа
В пробирку с притертой пробкой переносят 2 — 8 мл пробы, объем доводят до 8 мл поглотительным раствором, затем в пробирку вливают 1 мл раствора парарозанилина-хлоргидрата и 1 мл раствора формальдегида, содержимое пробирки встряхивают и через 20 мин. раствор переливают в кювету шириной 1 — 2 см и измеряют оптическую плотность по сравнению с контролем при длине волны 560 нм.
Окраска раствором устойчива в течение 30 мин.
Содержание сернистого газа в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 10).
Таблица 10
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
|
Поглотительный раствор, мл |
8 |
7,75 |
7,5 |
7,0 |
6,0 |
4,0 |
2,0 |
0 |
|
Содержание сернистого газа, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
8,0 |
12,0 |
16,0 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
Шкала может быть использована для визуального определения.
Примечание. Если в воздухе присутствовал сероводород, то он, соединяясь с солями ртути, образует нерастворимый осадок сульфида ртути. Последний удаляют фильтрованием или центрифугированием.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО АНГИДРИДА С ХЛОРИДОМ БАРИЯ [6]
Принцип определения
Сернистый газ окисляется хлоратом калия до серной кислоты, которая образует с хлоридом бария нерастворимый сульфат бария. Интенсивность помутнения пропорциональна концентрации сернистого газа.
Чувствительность определения — 3 мкг в анализируемом объеме раствора. Серная кислота и растворимые сульфаты мешают определению.
Реактивы
1. Поглотительный раствор. 4% раствор хлората калия: 4 г перекристаллизованного хлората калия растворяют в 100 мл дважды дистиллированной воды, в которой не содержится сульфатов.
2. Рабочий стандартный раствор сернистого газа готовят растворением 0,0272 г сульфата калия в воде, в мерной колбе емкостью 100 мл; 1 мл раствора соответствует 100 мкг сернистого газа.
3. Соляная кислота, 0,3% раствор.
4. Хлорид бария, 5% раствор.
5. Спирт этиловый, 96°.
6. Этиленгликоль.
7. Составной реактив. Готовят смешением трех объемов этиленгликоля с одним объемом 5% раствора хлорида бария и тремя объемами этилового спирта. Величину pH смеси доводят до 2,5 — 2,8 (проверяют на pH-метре). Составной реактив готовят за 2 дня до употребления. Годен к применению в течение 2 мес.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 2 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток протягивают воздух со скоростью 0, 25 л/мин. через поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически доводят до первоначального. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора доводят до первоначального объема бидистиллятом. 3 мл раствора помещают в пробирку, добавляют 2 мл составного реактива и осторожно перемешивают, избегая образования пузырьков. Через 5 мин. определяют интенсивность помутнения раствора в кюветах с толщиной слоя 1 см, при длине волны 410 или 540 нм по сравнению с контролем. Содержание сернистого газа в анализируемом объеме раствора определяют по заранее построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 11).
Таблица 11
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
|
Содержание сернистого газа, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
60 |
Все пробирки шкалы и пробы обрабатывают аналогично пробе, измеряют оптические плотности и строят график.
Шкала может быть использована для визуального определения.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СЕРНОЙ КИСЛОТЫ С ХЛОРИДОМ БАРИЯ [7]
Принцип определения
Серная кислота, взаимодействуя с хлоридом бария, образует сульфат бария, нерастворимый в воде. Интенсивность помутнения раствора пропорциональна количеству серной кислоты.
Чувствительность определения — 3 мкг серной кислоты в анализируемом объеме раствора. Определению мешают сульфаты, сернистый газ определению не мешает.
Реактивы
1. Исходный стандартный раствор готовят растворением 0,1776 г перекристаллизованного сернокислого калия в мерной колбе емкостью 100 мл в бидистиллированной воде. 1 мл раствора соответствует 1 мг серной кислоты.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разбавлением.
2. Реактивы — см. «Определение сернистого газа с хлоридом бария».
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА, укрепленный в патрон. Продолжительность отбора 20 мин.
б) Для определения среднесуточной концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА непрерывно в течение суток.
Допустимо среднесуточный отбор проводить на один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа
Вынимают фильтр из патрона, обрезают края и помещают в воронку с пористой пластинкой N 1. Воронку вставляют в пробирку с меткой на 10 мл, имеющую боковой отросток, который присоединен к водоструйному насосу. Наносят на фильтр 1 мл этанола и промывают его 4 — 5 раз горячим бидистиллятом. Доводят объем до 10 мл. Для контроля таким же образом промывают неиспользованный фильтр.
К 3 мл пробы добавляют 2 мл составного реактива и осторожно перемешивают, избегая образования воздушных пузырьков. Через 5 мин. определяют интенсивность помутнения раствора в кюветах с толщиной слоя 1 см, при длине волны 365 нм по сравнению с контролем. Если в пробе ожидается малое содержание серной кислоты, то пробу упаривают до объема 5 мл на кипящей водяной бане, для анализа берут также 3 мл. Содержание серной кислоты устанавливают по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 12).
Таблица 12
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
|
Содержание серной кислоты, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
60 |
80 |
Все пробирки шкалы и пробы обрабатывают аналогично пробе, измеряют оптическую плотность и строят калибровочный график.
Шкала может быть использована для визуального определения.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СЕРНИСТОГО ГАЗА И СЕРНОЙ КИСЛОТЫ [6]
Принцип определения
Аэрозоль серной кислоты отбирают на фильтр АФА. Сернистый газ фильтром не задерживается, а поглощается в поглотительном приборе Рыхтера, наполненном поглотительным раствором.
Серную кислоту определяют нефелометрическим методом, сернистый газ — колориметрическим или нефелометрическим методами.
Расчет см. выше.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 3 — 4 л/мин. протягивают через фильтр АФА, помещенный в патрон, к которому присоединен поглотительный прибор Рыхтера, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 3 — 4 л/мин. протягивают через аналогичную систему в течение суток. Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа см. выше.
ОПРЕДЕЛЕНИЕ СЕРНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА [8]
Принцип определения
Серная кислота окрашивает раствор индикатора водного голубого в синий цвет. Интенсивность окраски пропорциональна концентрации водородных ионов серной кислоты.
Чувствительность определения — 0,01 мкг водородных ионов в анализируемом объеме раствора.
Определению мешают аэрозоли щелочей. Сернистый газ определению не мешает.
Реактивы
1. Дистиллированная вода, дважды перегнанная, pH 5,4 — 6,6.
2. Исходный стандартный раствор серной кислоты. 0,005 н. раствор серной кислоты готовят соответствующим разбавлением 0,1 н. раствора серной кислоты. 1 мл раствора содержит 5 мкг водородных ионов. Рабочий стандартный раствор готовят разбавлением исходного раствора в 10 раз. 1 мл рабочего раствора содержит 0,5 мкг ионов водорода.
3. 0,02% раствор индикатора водного голубого в воде.
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 10 л/мин. протягивают через фильтр АФА, помещенный в патрон. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают через фильтр непрерывно в течение суток. Допустимо отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 2 — 4 ч.
Ход анализа
Фильтр вынимают из патрона, срезают края и помещают в воронку с пористой пластинкой N 1. Воронку вставляют в пробирку с меткой на 10 мл, имеющую боковой отросток, который присоединен к водоструйному насосу. Наливают на фильтр 1 мл этанола и промывают фильтр 4 — 5 раз горячей водой до общего объема 10 мл. Так же промывают неиспользованный фильтр для контроля.
Для анализа берут 5 мл промывной жидкости, добавляют 0,5 мл 0,02% раствора водного голубого и взбалтывают содержимое. Через 30 мин. измеряют интенсивность окраски раствора в кюветах с толщиной слоя 1 см при длине волны 592 нм по сравнению с контролем.
Содержание серной кислоты устанавливают по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 13).
Таблица 13
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
Поглотительный раствор, мл |
5 |
4,98 |
4,95 |
4,9 |
4,8 |
4,7 |
4,7 |
4,5 |
|
Содержание серной кислоты, мкг |
0 |
0,01 |
0,025 |
0,05 |
0,1 |
0,15 |
0,2 |
0,25 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график. Шкала может быть использована для визуального определения.
Расчет см. выше.
Примечание. Для выражения результатов анализа по молекуле серной кислоты надо провести пересчет. Коэффициент пересчета 49,05.
СЕРОВОДОРОД
Бесцветный газ, при большом разбавлении имеет неприятный запах, плотность по отношению к воздуху 1,1906, тяжелее воздуха. Вес 1 л газа 1,5392 г, он легко конденсируется в бесцветную жидкость. Сероводород растворим в воде. Один объем воды при 0 °C может поглотить 4,65, при 10 °C — 3,44 и при 20 °C — 2,61 объема сероводорода; он хорошо растворим в спирте: один объем спирта растворяет 11,8 объема при 10 °C. Сероводород является энергичным восстановителем, водный раствор его имеет слабокислую реакцию.
Сероводород — сильный нервный яд, небольшие концентрации которого действуют на слизистые оболочки дыхательных путей и глаз, вызывают головную боль, светобоязнь и боль в глазницах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРОВОДОРОДА С ПАРААМИНОДИМЕТИЛАНИЛИНОМ [4]
Принцип определения
При взаимодействии сероводорода с парааминодиметиланилином в кислой среде в присутствии трехвалентного железа образуется метиленовая синь. По интенсивности окраски раствора определяют содержание сероводорода.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Хлор, окислы азота мешают определению в концентрациях, в 5 раз превышающих сероводород, сернистый газ — в концентрациях, превышающих в 20 раз.
Реактивы
1а. Поглотительный раствор: 2,15 г сернокислого кадмия (CdSO4·8H2O) растворяют в 150 мл воды в колбе емкостью 500 мл и затем добавляют в колбу емкостью 100 мл 0,1 н. раствора едкого натра (постепенно, при взбалтывании), при этом образуется осадок гидроокиси кадмия. Раствор в колбе доводят водой до метки. При применении раствор хорошо взбалтывают. Эта суспензия применяется в качестве поглотительного раствора.
1б. В качестве поглотительного раствора можно применять 2% раствор ацетата цинка, подкисленного ледяной уксусной кислотой, 0,2 мл кислоты на 100 мл воды.
2. Стандартный раствор сероводорода. Исходный раствор готовят насыщением сероводородом 0,1 н. раствора едкого натра, приготовленного на дважды перегнанной воде.
Содержание сероводорода в 1 мл раствора устанавливают йодометрически. В коническую колбу вносят 10 мл приготовленной сероводородной воды, в которую предварительно было налито 20 мл 0,1 н. раствора йода, подкисленного 2 мл 5 н. соляной кислоты. Избыток йода, не связанный с сероводородом, оттитровывают 0,1 н. раствором тиосульфата в присутствии 0,5% раствора крахмала. 1 мл 0,1 н. раствора тиосульфата соответствуют 1,7043 мг сероводорода.
Для приготовления стандартного раствора А с содержанием 50 мкг/мл в мерную колбу емкостью 100 мл вносят такое количество исходного раствора, в котором содержится 5 мг сероводорода, и колбу доливают дважды перегнанной водой до метки.
Содержание сероводорода в 1 мл устанавливают йодометрически, титруя раствор 0,005 н. тиосульфатом, 1 мл которого соответствует 85,20 мкг сероводорода.
Рабочий стандартный раствор с содержанием 2 мкг/мл готовят из раствора А в мерной колбе емкостью 100 мл, в которую вливают количество раствора А, содержащее 200 мкг сероводорода. Перед приготовлением рабочего стандартного раствора раствор А необходимо титровать.
Рабочий стандартный раствор устойчив в течение 40 — 50 мин.
3. Парааминодиметиланилин сульфат, 0,1% раствор: 0,05 г растворяют в колбе емкостью 50 мл в 25 мл соляной кислоты, уд. вес 1,19, и добавляют водой до метки. Сохраняется в течение 2 нед. в холодном темном месте.
4. Хлорное железо, 0,12% раствор. 0,12 г трехвалентного хлорного железа растворяют в 10 мл соляной кислоты, уд. вес 1,19, и объем раствора доводят до 100 мл водой.
5. Едкий натр, 0,1 н. раствор.
6. Тиосульфат, 0,1 н. раствор. Тиосульфат 0,005 н. раствор готовят перед применением из 0,1 н. раствора.
7. Йод, 0,1 н. раствор.
8. Сорбент для задержания сернистого ангидрида. Пемзу с размером частиц 3 — 5 мм пропитывают насыщенным раствором ацетата натрия, высушивают в сушильном шкафу при 110 — 120 °C и наполняют стеклянный патрон (диаметр 2 см), в который насыпают 8 — 10 см3 сорбента. Патрон соединяют с входным отверстием поглотительного прибора, помещая его в вертикальном положении.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 2 л/мин. через два поглотительных прибора с пористым фильтром N 1, содержащие по 1 мл поглотительного раствора и 4 мл воды. Продолжительность отбора 20 — 30 мин. Поглотительный прибор должен быть защищен от света.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 25 л/ч протягивают через два поглотительных прибора с пористым фильтром N 1, содержащих по 1 мл поглотительного раствора и 7 мл воды.
При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Прибор защищают от света.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 — 2 ч в один и тот же прибор.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. Раствор из поглотительного прибора переносят в мерную колбу емкостью 10 мл или в пробирку с меткой 10 мл и с притертой пробкой, добавляют 0,5 мл раствора парааминодиметиланилина, 0, 5 мл раствора хлорного железа и через 10 мин. доливают водой до метки. Через 10 мин. раствор переводят в кювету шириной 2 см и измеряют оптическую плотность по сравнению с контролем, который готовят одновременно и аналогично пробе при длине волны 665 нм.
Содержание сероводорода в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов в колбах емкостью 10 мл (табл. 14).
Таблица 14
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
|
Поглотительный раствор, мл |
По 1 мл в каждую колбу |
|||||
|
Содержание сероводорода, мкг |
0 |
1 |
2 |
3 |
4 |
5 |
Объем растворов доводят до 10 мл и далее обрабатывают аналогично пробе.
Измеряют оптическую плотность и строят калибровочный график.
Для визуального определения готовят шкалу согласно табл. 15.
Таблица 15
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,15 |
0,25 |
0,5 |
1 |
1,5 |
2 |
|
Поглотительный раствор, мл |
По 1 мл |
||||||
|
Вода, мл |
4 |
3,85 |
3,75 |
3,5 |
3 |
2,5 |
2 |
|
Содержание сероводорода, мкг |
0 |
0,3 |
0,5 |
1 |
2 |
3 |
4 |
Для анализа берут всю пробу, переносят в колориметрические пробирки, вносят по 0,25 мл раствора парааминодиметиланилина и по 0,25 мл раствора хлорного железа. Через 10 мин. проводят определение.
Расчет см. выше.
Примечания: 1. Для анализа, кроме сульфата парааминодиметиланилина, можно использовать и хлоргидрат, приготовленный следующим образом. Сначала готовят 0,5% раствор метилового оранжевого. Для этого навеску 0,5 г растворяют в воде в мерной колбе емкостью 100 мл при нагревании (50 — 60 °C). После охлаждения до комнатной температуры раствор доводят водой до метки. Перед применением раствор слегка нагревают для растворения выпавших кристаллов.
К 10 мл 0,5% раствора метилового оранжевого прибавляют 10 мл концентрированной соляной кислоты, перемешивают и добавляют для восстановления 5 г порошкообразного металлического цинка, который прибавляют небольшими порциями до обесцвечивания раствора. Последнее происходит через 15 — 20 мин. Затем цинк быстро отфильтровывают через вату, промывают 10 мл воды, к прозрачному раствору прибавляют еще 15 мл концентрированной соляной кислоты и доводят объем водой до 50 мл.
Реактив хранят в темном и холодном месте, его можно использовать до тех пор, пока он бесцветный или бледно-желтого цвета. Перед употреблением реактив следует проверять на стандартных растворах.
2. Отобранные пробы хранят в темном и холодном месте, их анализ рекомендуется производить в течение суток.
ОПРЕДЕЛЕНИЕ СЕРОВОДОРОДА С НИТРАТОМ СЕРЕБРА [5]
Принцип определения
При взаимодействии сероводорода с нитратом серебра образуется коллоидальный раствор сульфида серебра, окрашенный в желто-коричневый цвет. По интенсивности окраски раствора определяют содержание сероводорода.
Чувствительность определения 1 мкг в анализируемом объеме раствора.
Мешают определению галоиды.
Реактивы
1. Поглотительный раствор готовят из ортоарсенита натрия или мышьяковистого ангидрида.
В первом случае растворяют 2 г арсенита натрия в 100 мл 5% раствора карбоната аммония и объем раствора доводят до 1 л водой.
Во втором случае 2 г мышьяковистого ангидрида и 5 г карбоната аммония вносят в небольшой объем воды, подогревают до растворения ангидрида и доводят объем раствора до 1 л водой.
2. Стандартный раствор сероводорода. Исходный раствор с содержанием 100 мкг/мл готовят следующим образом: вливают 3 мл 0,1 н. раствора тиосульфата в мерную колбу емкостью 100 мл и добавляют до метки 0,5% раствором карбоната аммония.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз поглотительным раствором. Готовят перед анализом.
3. Нитрат серебра (1% раствор): 1 г нитрата серебра растворяют в 80 мл воды и осторожно добавляют 10 мл концентрированной серной кислоты. После охлаждения раствор доливают водой до 100 мл.
4. Карбонат аммония (0,5% раствор).
5. Крахмал (1% раствор).
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 4 л/мин. протягивают через поглотительный прибор Рыхтера, содержащий 6 мл поглотительного раствора.
Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации воздух непрерывно в течение суток со скоростью 25 л/ч протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального уровня.
Допустимо проводить отбор с перерывами в 2 — 4 ч в один и тот же прибор 6 — 12 раз.
Ход анализа
В колориметрическую пробирку переносят 5 мл пробы. Одновременно готовят шкалу стандартов (табл. 16).
Таблица 16
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание сероводорода, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Во все пробирки шкалы и проб наливают по 0,2 мл крахмала и по 1 мл нитрата серебра. Содержимое пробирок встряхивают и через 5 мин. производят сравнения интенсивности окраски проб со шкалой стандартов.
Расчет см. выше.
Примечание. Определение можно проводить фотометрически при длине волны 346 нм в кювете с толщиной слоя 1 — 2 см. Чувствительность определения 5 мкг в анализируемом объеме раствора.
СЕРОУГЛЕРОД
Бесцветная жидкость, в чистом виде имеет приятный запах, но при наличии примесей обычно обладает неприятным запахом. Удельный вес при 25 °C 1,263, кипит при 46,3 °C. Пары чрезвычайно легко воспламеняются, они тяжелее воздуха в 2,6 раза. Сероуглерод нерастворим в воде, но хорошо растворяется в ряде органических растворителей; он является хорошим растворителем для жиров, масел, смол, каучука, серы, фосфора, йода и некоторых других трудно растворимых веществ.
Сероуглерод токсичен, действует на центральную нервную систему, поражает и внутренние органы.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ СЕРОУГЛЕРОДА С ДИЭТИЛАМИНОМ [2]
Принцип определения
Сероуглерод реагирует с диэтиламином, образуя диэтилдитиокарбамат, который при наличии ионов двухвалентной меди образует желто-коричневую окраску вследствие образования комплексной соли. Интенсивность окраски раствора пропорциональна количеству сероуглерода.
Чувствительность: 1 мкг CS2 в определяемом объеме.
Определению мешает сероводород.
Влияние сероводорода устраняют применением предохранительного фильтра. Меркаптаны, тиофен, диметилсульфид определению не мешают.
Реактивы
1. Поглотительный раствор: 0,1 г ацетата меди х.ч. (CH3COO)·CuH2O растворяют в 5 мл воды и 10 мл дважды перегнанного триэтаноламина, прибавляют 5 мл чистого диэтиламина (или 15 мл 33% раствора) и доводят этанолом до 1 л. Можно пользоваться также этанолом, содержащим 1% бензина. Если приготовленный поглотительный раствор слегка опалесцирует, ему дают перед применением отстояться в течение 2 — 3 дней. Реактив в хорошо закрытой бутылке устойчив в течение нескольких недель.
2. Стандартный раствор сероуглерода. Исходный раствор готовят в мерной колбе емкостью 50 мл, в которую вливают 10 — 15 мл этанола. Колбу взвешивают и вносят в нее 2 — 3 капли свежеперегнанного сероуглерода и вновь взвешивают. Объем жидкости в колбе доводят до метки этанолом и рассчитывают количество сероуглерода в 1 мл.
Рабочий стандартный раствор с содержанием сероуглерода 2 мкг/мл получают в мерной колбе емкостью 50 мл, в которую предварительно вливают 15 — 20 мл этанола, а затем вносят такое количество исходного раствора, которое содержит 100 мкг сероуглерода, и раствор доводят до метки этанолом.
3. Вата для улавливания H2S [3].
В 50 мл воды, подкисленной несколькими каплями уксусной кислоты, растворяют 10 г ацетата свинца, прибавляют 10 мл глицерина и доводят водой до 100 мл. Хлопчатобумажную вату, пропитанную этим раствором, выжимают, высушивают при комнатной температуре и хранят в банке с притертой пробкой.
Отбор пробы
а) Для определения разовой концентрации исследуемый воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор, наполненный 5 мл поглотительного раствора. К поглотителю присоединена стеклянная трубочка, наполненная ватой для улавливания сероводорода.
Продолжительность отбора 20 — 30 мин. при охлаждении.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 7,5 л/ч протягивают через поглотительный прибор, содержащий 6 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют этанолом до первоначального 33% раствором диэтиламина.
Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора переносят в пробирку емкостью 10 мл и доводят до метки поглотительным раствором. Интенсивность окраски измеряют через 15 мин. с помощью фотометра в кювете шириной от 1 до 5 см при длине волны 465 нм по сравнению с контролем (поглотительным раствором). Окраска устойчива в течение суток.
Содержание сероуглерода в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 17).
Таблица 17
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
6,0 |
7,0 |
8,0 |
10 |
|
Содержание сероуглерода, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
12 |
14 |
16 |
20 |
Объем каждой пробирки доводят до метки поглотительным раствором. Измеряют оптическую плотность и строят калибровочный график в координатах концентрация — оптическая плотность. Шкала может быть использована для визуального определения.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеев М.В., Самородина Р.Я. Колориметрическое определение содержания сернистого газа в атмосферном воздухе. Гиг. и сан., 1953, 10.
2. Определение сернистого газа по реакции с фуксином. Определение сернистого газа по реакции с парарозанилином. Определение сероуглерода. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
3. Павленко А.А., Кузьмина Т.А. Предохранительный патрон для задержания сероводорода, сернистого газа. Труды Главной геофизической обсерватории. Л., 1968, в. 234.
4. Пауль И.И., Виноградова В.А. Определение малых количеств сероводорода в воздухе. Гигиена труда, 1960, 2.
5. Полежаев Н.Г. К методике определения малых количеств сероводорода в воздухе. Гигиена труда и техника безопасности, 1937, 6.
6. Чепкевичене Е.С. Разработка методов определения серной кислоты и раздельного определения серной кислоты, сернистого ангидрида и сульфатов в воздухе при гигиенических исследованиях. Автореф. дисс. канд. М., 1972.
7. Чепкевичене Е.С. К вопросу определения аэрозоля серной кислоты в атмосферном воздухе. Вопросы эпидемиологии и гигиены в Литовской ССР. Вильнюс, 1969.
8. Чепкевичене Е.С. Определение серной кислоты по водородному катиону. Вопросы эпидемиологии и гигиены в Литовской ССР. Вильнюс, 1971.
Глава IV
СОЕДИНЕНИЯ АЗОТА
АММИАК
Бесцветный газ с резким запахом, хорошо растворимый в воде, в спирте растворяется 13,2% при 20 °C.
В продаже бывает в виде водных растворов с содержанием 28 — 29% аммиака (удельный вес 0,90 при 25 °C). Плотность аммиака по отношению к воздуху 0,59.
Температура плавления 77,7 °C, температура кипения 33,35 °C. Вес 1 л газообразного аммиака при нормальных условиях 0,771 г.
Аммиак оказывает раздражающее действие на верхние дыхательные пути, а также на слизистые оболочки глаз.
Предельно допустимые концентрации аммиака; максимальная разовая 0,20 мг/м3, среднесуточная 0,20 мг/м3.
ОПРЕДЕЛЕНИЕ АММИАКА С РЕАКТИВОМ НЕССЛЕРА [2]
Принцип определения
При взаимодействии аммиака с реактивом Несслера образуется соединение, окрашенное в желто-бурый цвет. Интенсивность окраски пропорциональна количеству иона аммония.
Чувствительность определения 2 мкг аммиака в анализируемом объеме раствора.
Определению мешают аммонийные соли, некоторые амины алифатического ряда, формальдегид с 0,2 мг/м3, сероводород с 0,04 мг/м3, хлор с 0,2 мг/м3. Окислы азота определению не мешают.
Реактивы
Все реактивы готовят на дистиллированной воде, не дающей положительной реакции на ион ![]()
1. Поглотительный раствор: серная кислота 0,01 н. раствор.
2. Стандартный раствор.
Исходный раствор с содержанием 100 мкг/мл аммиака готовят растворением 0,3140 г хлорида аммония в 100 мл дистиллированной воды.
Раствор устойчив в течение 1 — 2 мес.
Рабочий стандартный раствор, содержащий 10 мкг/мл аммиака, готовят разведением исходного раствора 100 раз водой. Раствор готовят перед анализом.
3. Реактив Несслера.
а) 17 г хлорида ртути растворяют в 300 мл воды, в другой колбе растворяют 35 г йодида калия в 100 мл воды. Ко второму раствору приливают первый до образования красного нерастворимого осадка, затем добавляют 600 мл 20% раствора едкого натра и оставшееся количество раствора хлорида ртути, раствору дают отстояться в темном месте, после чего осторожно сифонируют в темную склянку и закрывают резиновой пробкой.
При отстаивании в реактиве может образоваться осадок, но если жидкость над осадком слегка желтоватого цвета, прозрачный раствор пригоден к употреблению.
б) 10 г йодида ртути растирают в ступке с небольшими количествами воды (около 10 мл), переносят эту взвесь в склянку из темного стекла, ступку ополаскивают небольшим количеством воды, сливая ее в ту же склянку, и прибавляют 5 г йодида калия и 40 г едкого натра, растворенного в 60 — 70 мл дистиллированной воды. Реактив оставляют на 1 — 2 дня, после чего осторожно сифонируют прозрачный раствор в склянку из темного стекла и закрывают резиновой пробкой.
4. Дистиллированная вода, не дающая положительной реакции на ион ![]() Для ее получения к 1 л дистиллированной воды прибавляют 5 мл 10% серной кислоты и перегоняют ее. Первые порции погона (100 — 200 мл) отбрасывают. Следующий погон проверяют с реактивом Несслера на содержание иона
Для ее получения к 1 л дистиллированной воды прибавляют 5 мл 10% серной кислоты и перегоняют ее. Первые порции погона (100 — 200 мл) отбрасывают. Следующий погон проверяют с реактивом Несслера на содержание иона ![]() При отрицательной реакции воду собирают и хранят в закрытой бутыли.
При отрицательной реакции воду собирают и хранят в закрытой бутыли.
Безаммиачную воду можно получать также при пропускании дистиллированной воды со скоростью 5 мл в мин. через колонку с катионитами КУ-2 или СБС. Предварительно проводят подготовку катионита. С этой целью катионит помещают в стакан и заливают дистиллированной водой. На следующий день катионит многократно обрабатывают 5% раствором соляной кислоты для освобождения его от железа, проверяют присутствие его качественной реакцией с роданистым аммонием. Далее катионит переносят в бюретку емкостью 50 — 100 мл, смывая его дистиллированной водой. В нижнюю часть бюретки предварительно помещают слой стеклянной ваты толщиной 1 — 2 см.
Соляную кислоту пропускают через катионит медленно, до отрицательной реакции на железо. Подготовка катионита заканчивается промыванием его дистиллированной водой до нейтральной реакции.
Катионит следует хранить под слоем дистиллированной воды.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через 2 поглотительных прибора с пористой пластинкой N 1, содержащих по 6 мл 0,01 н. раствора серной кислоты. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,10 — 0,2 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 6 мл 0,01 н. раствора серной кислоты, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
Из каждого поглотительного прибора 5 мл пробы вносят в колориметрические пробирки, приливают по 0,5 мл реактива Несслера, взбалтывают и через 5 — 10 мин. фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 450 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание аммиака в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 18).
Таблица 18
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,6 |
4,2 |
4,0 |
3,0 |
|
Содержание аммиака, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
Примечание. Реактив Несслера следует вливать быстро, тогда устраняется опалесценция раствора.
ОПРЕДЕЛЕНИЕ АММИАКА С НАТРИЕВОСАЛИЦИЛАТНЫМ РЕАКТИВОМ [2]
Принцип определения
Метод основан на образовании соединения, окрашенного в зеленовато-синий цвет при взаимодействии аммиака с натриевосалицилатным реактивом в присутствии гипохлорита натрия.
Чувствительность определения 0,1 мкг аммиака в анализируемом объеме раствора.
Сернистый газ в концентрации 3 мг/м3, сероводород — 0,2 мг/куб. м, сероуглерод — 0,7 мг/куб. м, фенол — 0,1 мг/м3 и альдегиды не мешают определению.
Реактивы
1. Поглотительный раствор: 0,02 н. раствор серной кислоты.
2. Исходный стандартный раствор с содержанием 100 мкг/мл аммиака готовят растворением 0,0314 г хлорида аммония в дважды перегнанной воде, не содержащей аммиака.
Рабочий стандартный раствор с содержанием 1 мкг/мл аммиака готовят разведением исходного раствора поглотительным раствором.
3. Едкий натр.
4. Хлорная вода (0,5 — 0,6% раствор).
5. Гипохлоритный реактив. 16 г едкого натра растворяют в 100 мл 0,5 — 0,6% хлорной воды в мерной колбе емкостью 100 мл.
6. Салицилат натрия.
7. Нитропруссид натрия.
8. Натриевосалицилатный реактив. К 45 г салицилата натрия прибавляют 50 мг нитропруссида натрия, предварительно хорошо растертого в ступке с 73 мл дважды перегнанной воды. Реактив сохраняют в темном полиэтиленовом сосуде, в темноте. При комнатной температуре реактив устойчив в течение 5 мес.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. непрерывно в течение суток протягивают через поглотительный прибор с пористым фильтром N 1, содержащий 6 мл поглотительного раствора. Допустимо проводить отбор 6 — 12 раз с перерывом в 4 — 2 ч в один и тот же поглотительный прибор.
Ход анализа
Содержимое поглотительного прибора доводят до объема 6 мл поглотительным раствором, 5 мл раствора берут на анализ, добавляют 0,5 мл натриево-салицилатного реактива, взбалтывают, приливают 0,5 мл гипохлоритного реактива и снова взбалтывают. Через 2 1/2 — 3 ч фотометрируют в кюветах с толщиной слоя 1 см при длине волны 597 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание аммиака в анализируемом объеме определяют по градуировочному графику, который предварительно строят по шкале стандартов (табл. 19).
Таблица 19
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание аммиака, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения, готовят ее одновременно с пробами в колориметрических пробирках.
Расчет количества аммиака см. выше.
Примечания. 1. Все реактивы готовят на дважды перегнанной безаммиачной воде. Приготовление безаммиачной воды см. выше.
2. Так как описанный метод определения аммиака очень чувствителен, посуду необходимо тщательно промывать горячей хромовой смесью, водой, 20% раствором едкого натра и еще раз водой до нейтральной реакции.
ДВУОКИСЬ АЗОТА
|
NO2 |
Мол. вес 46,01 |
|
N2O4 |
Мол. вес 92,02 |
Двуокись азота — желто-бурый газ с резким запахом, легко сгущающийся в жидкость, кипящую при +21 °C, при охлаждении до -11 °C эта жидкость застывает в бесцветную кристаллическую массу. При температуре ниже 140 °C двуокись азота частично полимеризуется в N2O4, и около точки замерзания (-11 °C) вещество состоит только из молекул N2O4. При повышении температуры молекула N2O4 диссоциирует на две молекулы NO2. При растворении NO2, N2O4 в воде образуются азотистая и азотная кислоты.
Двуокись азота раздражающе действует на легкие, вызывая в тяжелых случаях отек их, на верхние дыхательные пути и глаза, а также оказывает общетоксическое действие.
Предельно допустимые концентрации двуокиси азота: максимальная разовая и среднесуточная 0,085 мг/м3.
ОПРЕДЕЛЕНИЕ ДВУОКИСИ АЗОТА [3] С РЕАКТИВОМ ГРИССА — ИЛОСВАЯ
Принцип определения
Метод основан на поглощении двуокиси азота в раствор йодида калия и определения иона нитрита по реакции Грисса — Илосвая.
Чувствительность определения 0,3 мкг двуокиси азота в анализируемом объеме раствора.
Озон не мешает определению до концентраций, превышающих концентрации двуокиси азота в 2 — 3 раза.
Реактивы
1. Поглотительный раствор: 0,5 н. раствор йодида калия.
2. Стандартные растворы нитрита натрия. Исходный стандартный раствор с содержанием 1000 мкг/мл иона ![]() готовят растворением 0,150 г перекристаллизованного нитрита натрия в 100 мл дважды перегнанной воды. Рабочий стандартный раствор с содержанием 1 мкг/мл иона
готовят растворением 0,150 г перекристаллизованного нитрита натрия в 100 мл дважды перегнанной воды. Рабочий стандартный раствор с содержанием 1 мкг/мл иона ![]() готовят разведением исходного раствора в 1000 раз поглотительным раствором.
готовят разведением исходного раствора в 1000 раз поглотительным раствором.
3. Сульфит натрия, 0,01 н. раствор: 0,0630 г безводного сульфита или 0,1261 г Na2SO3·7H2O растворяют в 100 мл дистиллированной воды. Раствор готовят перед анализом.
4. Уксусная кислота (12% раствор).
5. Раствор сульфаниловой кислоты. 0,5 г сульфаниловой кислоты растворяют в 150 мл 12% раствора уксусной кислоты.
6. Раствор ![]() -нафтиламина. 0,2 г
-нафтиламина. 0,2 г ![]() -нафтиламина кипятят с 20 мл воды в течение 2 — 3 мин. до образования на дне капли лилового цвета. Бесцветный раствор декантируют и вливают в 150 мл 12% раствора уксусной кислоты.
-нафтиламина кипятят с 20 мл воды в течение 2 — 3 мин. до образования на дне капли лилового цвета. Бесцветный раствор декантируют и вливают в 150 мл 12% раствора уксусной кислоты.
Растворы сульфаниловой кислоты и ![]() -нафтиламина хранят в темных склянках. Они пригодны к употреблению до тех пор, пока не побуреют.
-нафтиламина хранят в темных склянках. Они пригодны к употреблению до тех пор, пока не побуреют.
7. Реактив Грисса — Илосвая. Перед употреблением смешивают растворы сульфаниловой кислоты и ![]() -нафтиламина в отношении 1:1.
-нафтиламина в отношении 1:1.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1 с 6 мл поглотительного раствора в каждом.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,05 л/мин. (3 л/ч) протягивают через два поглотительных прибора, содержащих по 6 мл поглотительного раствора в каждом, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В колориметрические пробирки переносят 5 мл пробы из каждого поглотительного прибора, приливают по 0,5 мл реактива Грисса — Илосвая и хорошо перемешивают. Через 10 мин после внесения реактива Грисса — Илосвая непосредственно перед определением добавляют 5 капель 0,01 н. раствора сульфита натрия, взбалтывают и сразу фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 520 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание двуокиси азота в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 20).
Таблица 20
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,5 |
2 |
|
Поглотительный раствор, мл |
5 |
4,7 |
4,5 |
4,3 |
4,1 |
3,6 |
3 |
|
Содержание двуокиси азота, мкг |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,5 |
2 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят калибровочный график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами в объеме 2 мл раствора.
Чувствительность определения — 0,1 мкг в анализируемом объеме раствора. В этом случае отбор проб проводят в 2 — 4 мл поглотительного раствора.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ ДВУОКИСИ АЗОТА С ЗАМЕЩЕННЫМ ЭТИЛЕНДИАМИНОМ [2]
Принцип определения
При взаимодействии двуокиси азота в кислой среде с сульфаниловой кислотой образуется диазосоль, которая дает с N-(1-нафтил)-этилендиамином красную окраску, интенсивность которой пропорциональна количеству двуокиси азота.
Чувствительность определения: 0,05 мкг двуокиси азота в 1 мл исследуемого раствора.
Определению мешают сероводород с 0,05 мг/м3 , сернистый газ с 0,4 мг/куб. м, хлор с 0,02 мг/куб. м, формальдегид с 0,08 мг/м3. Закись азота, трехокись азота и нитраты определению не мешают.
Реактивы
1. Поглотительный раствор: 2 мл 0,1% водного раствора HN-(1(нафтил-)-этилендиаминдихлоргидрата доводят в мерной колбе до 100 мл раствором сульфаниловой кислоты. Реактив должен быть бесцветный, его готовят перед анализом.
2. N-(1-нафтил-)-этилендиаминдихлоргидрат, 0,1% водный раствор.
При хранении в темном и прохладном месте устойчив в течение 2 нед.
3. Раствор сульфаниловой кислоты. 5 г сульфаниловой кислоты растворяют в 800 мл дистиллированной воды, подкисляют 140 мл ледяной уксусной кислоты и доводят до 1 л. Раствор следует хранить в темном и прохладном месте, тогда он устойчив в течение 2 нед.
4. Стандартные растворы нитрита натрия.
Исходный стандартный раствор с содержанием 1000 мкг/мл готовят путем растворения 0,3750 г нитрита натрия в 250 мл дистиллированной воды. В темноте и на холоде раствор устойчив в течение 2 нед.
Рабочий стандартный раствор с содержанием 5 мкг/мл. В мерной колбе емкостью 1000 мл разбавляют 5 мл исходного раствора дважды перегнанной водой.
Раствор должен быть свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два поглотительных прибора со стеклянным фильтром, наполненных по 5 мл поглотительного раствора. Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2,5 л/ч протягивают через два поглотительных прибора, содержащих по 5 мл поглотительного раствора, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывами 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
Содержимое каждого поглотителя доливают дистиллированной водой до прежнего объема и измеряют интенсивность окраски раствора на фотометре в кювете шириной от 1 до 5 см с зеленым светофильтром, с максимумом пропускания 550 нм до сравнению с поглотительным раствором. По калибровочному графику определяют содержание двуокиси азота в 2 мл раствора (табл. 21).
Таблица 21
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1,0 |
1,5 |
2,5 |
|
Поглотительный раствор, мл |
25 |
24,75 |
24,5 |
24,0 |
23,5 |
22,5 |
|
Содержание двуокиси азота, мкг/мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,5 |
Интенсивность окраски измеряют через 30 мин. с помощью фотометра в кюветах шириной 1 — 5 см с зеленым светофильтром — с максимумом пропускания 550 нм по сравнению с поглотительным раствором. Найденные величины оптической плотности помещают в графике напротив величин содержания двуокиси азота (мкг/мл).
Расчет анализа: <1>
———————————
<1> При расчете анализа надо учитывать, что концентрация двуокиси азота найдена в 1 мл раствора.
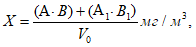
где X — концентрация искомого вещества в миллиграммах на 1 м3 воздуха; A — количество вещества в первом поглотительном приборе в микрограммах; A1 — количество вещества во втором поглотительном приборе в микрограммах; B — объем всей пробы в первом поглотительном приборе в миллиграммах; B1 — объем всей пробы во втором поглотительном приборе в миллиграммах; V0 — объем протянутого воздуха, приведенного к нормальным условиям в литрах.
Примечания. 1. При отборе пробы воздуха поглотительный раствор приобретает красную окраску, пропорциональную количеству двуокиси азота. Второй поглотитель — контрольный. Если наблюдается «проскок» во второй поглотитель, надо уменьшить скорость протягивания воздуха и добавить две капли бутанола.
2. Образующаяся окраска в темноте при комнатной температуре устойчива в течение 48 ч.
3. При использовании кювет с шириной менее 5 см соответственно нужно увеличить концентрацию растворов при построении градуировочной кривой.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ ОКИСИ И ДВУОКИСИ АЗОТА
Принцип определения [1]
Метод основан на окислении окиси азота до двуокиси с помощью окислительной смеси, нанесенной на твердый сорбент, и дальнейшем определении двуокиси азота по образованию азокрасителя с реактивом Грисса — Илосвая.
Чувствительность определения 0,3 мкг в анализируемом объеме раствора.
Озон не мешает определению двуокиси азота до концентраций, превышающих концентрации двуокиси азота в 2 — 3 раза.
Аппаратура
1. V-образная стеклянная трубка, d = 15 — 17 мм, высота — 10 — 12 см.
2. Поглотители с пористой пластинкой N 1.
Реактивы
1. 2,5% раствор серной кислоты.
2. Окислительная смесь — 3% раствор бихромата калия в 2,5% растворе серной кислоты.
3. Твердый сорбент:
а) Инзенский диатомит, размер зерен 0,1 — 0,2 мм, обработанный кипячением в течение 20 — 30 мин. в воде и высушенный на воздухе.
б) Стеклянный порошок, размер зерен 0,25 — 0,5 мм, обработанный кипячением в течение 1 — 1 1/2 ч с соляной кислотой (1:3), промытый водой и высушенный.
4. Твердый сорбент, пропитанный окислительной смесью.
8 — 10 г диатомита или 24 — 25 г стеклянного порошка помещают в фарфоровую чашку, заливают 30 мл окислительной смеси и оставляют на 2 ч. По истечении этого времени раствор сливают, сорбент подсушивают между листами фильтровальной бумаги, сушат в течение 15 — 20 мин. в сушильном шкафу при 50 — 60 °C и досушивают на воздухе. Сорбент, пропитанный окислительной смесью, хранят в склянке с притертой пробкой. Его можно хранить в течение длительного времени.
5. V-образная трубка с твердым сорбентом. V-образную трубку наполняют 6 — 8 г диатомита или 24 — 25 г стеклянного порошка, пропитанного окислительной смесью. В концы трубки вводят тампончики из ваты. Сопротивление трубок должно быть не менее 180 мм вод. ст. Трубка может быть использована на 5 — 6 ч работы.
Приготовление остальных реактивов — см. Определение двуокиси азота выше.
Отбор пробы
Исследуемый воздух со скоростью 0,1 — 0,2 л/мин. протягивают через систему, состоящую из двух поглотительных приборов с пористой пластинкой N 1, содержащих по 5 мл 0,5 н. раствора йодистого калия, и V-образной трубки с твердым сорбентом, пропитанного окислительной смесью (рис. 19). I — поглотительный прибор с 0,5 н. раствором йодистого калия, служащий для поглощения двуокиси азота из воздуха. II — V-образная трубка с твердым сорбентом, пропитанным окислительной смесью, служит для окисления окиси азота до двуокиси. III — поглотительный прибор с 0,5 н. раствором йодистого калия служит для поглощения двуокиси азота, образовавшейся в результате окисления окиси азота.
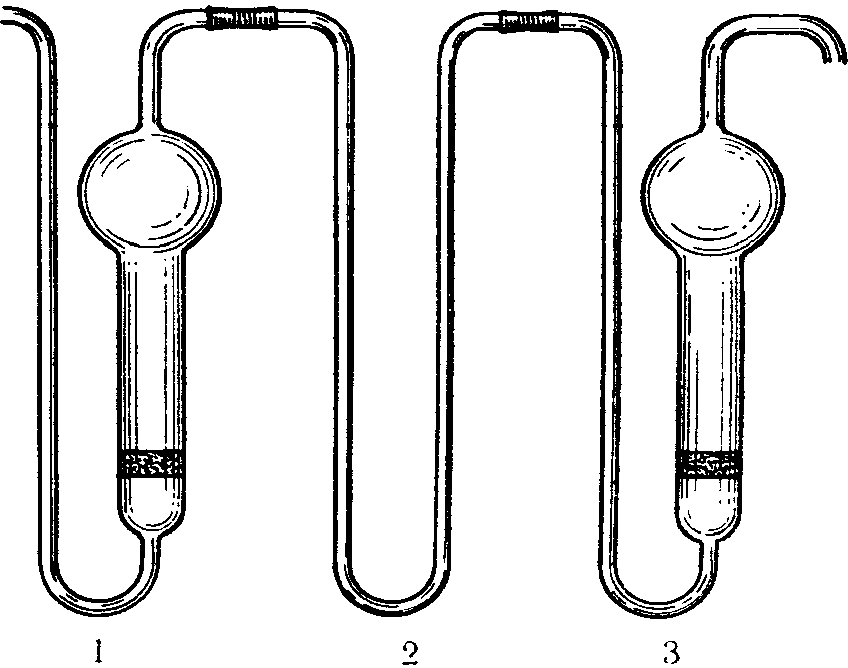
Рис. 19. Схема присоединения поглотительных
приборов при определении окислов азота
Ход анализа
В колориметрическую пробирку переносят 5 мл пробы, приливают 0,5 мл реактива Грисса — Илосвая и хорошо перемешивают. Через 10 мин. после внесения реактива Грисса — Илосвая непосредственно перед определением добавляют 5 капель 0,01 н. раствора сульфита натрия, взбалтывают и сразу фотометрируют в кюветах с толщиной слоя 1 см при длине волны 520 нм по сравнению с контролем. Содержание двуокиси азота в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику.
Для построения градуировочного графика готовят шкалу стандартов (табл. 22).
Таблица 22
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,2 |
1,5 |
|
Поглотительный раствор, мл |
5 |
4,7 |
4,5 |
4,3 |
4,1 |
3,8 |
3,5 |
|
Содержание двуокиси азота, мкг |
0 |
0,3 |
0,5 |
0,7 |
0,9 |
1,2 |
1,5 |
Градуировочный график. Шкалу стандартов обрабатывают аналогично пробам и измеряют оптическую плотность. График строят в координатах «концентрация — оптическая плотность». Шкалу стандартов можно использовать для визуального определения.
Расчет анализа
V — общий объем поглотительного раствора в первом поглотительном приборе (мл).
V1 — объем пробы, взятой для анализа из первого поглотительного раствора (мл).
а — количество двуокиси азота в определяемом объеме в микрограммах.
V0 — объем пробы воздуха в литрах (0°, 760 мм рт. ст.).
С1 — концентрация двуокиси азота в первом поглотительном приборе:
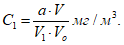
C2 — концентрация двуокиси азота во втором поглотительном приборе рассчитывается по этой же формуле.
Для получения суммы окиси и двуокиси азота (C ) количества двуокиси азота в первом и во втором поглотительных приборах суммируют:
C3 = C1 + C2 мг/м3.
Примечания. 1. При содержании в воздухе больших количеств двуокиси и окиси азота поглощение ведут не в один, а в два поглотительных прибора с йодидом калия.
2. При большой влажности воздуха перед системой ставят трубочку с пемзой, пропитанной серной кислотой.
АЗОТНАЯ КИСЛОТА
Бесцветная жидкость с едким запахом, уд. вес 1,52 (15 °C), температура кипения 86 °C, кипение сопровождается частичным разложением; пары HNO3 в 2,2 раза тяжелее воздуха. Смешивается с водой во всех отношениях, водные растворы полностью диссоциированы. Весьма гигроскопична, сильно дымит на воздухе. HNO3 — сильная кислота, действует на все металлы, кроме золота, платины, иридия и радия. Является сильным окислителем.
Действует раздражающе на дыхательные пути, может вызывать поражения роговицы глаз, разрушение зубов, при попадании на кожу вызывает тяжелые ожоги.
Предельно допустимые концентрации:
а) по молекуле HNO3: максимальная разовая 0,4 мг/м3, среднесуточная 0,4 мг/м3;
б) по водородному иону: максимальная разовая 0,006 мг/м3, среднесуточная 0,006 мг/м3.
ОПРЕДЕЛЕНИЕ АЗОТНОЙ КИСЛОТЫ ПО ИОНУ ВОДОРОДА
Принцип тот же, что и при определении соляной кислоты. Диссоциация азотной кислоты равнозначна соляной кислоте. Определение ее проводят аналогично соляной кислоте (см. выше).
ЛИТЕРАТУРА
1. Дмитриев М.Т., Соловьева Т.В. Раздельное определение окиси и двуокиси азота. Гиг. и сан., 1971, 9.
2. Определение аммиака по реакции с натриевосалицилатным реактивом. Определение аммиака с реактивом Несслера. Определение двуокиси азота с замещенным этилендиамином. В сб.: СЭВ. Унифицированные методы определения атмосферных загрязнений. М., 1970.
3. Полежаев Н.Г., Гирина В.В. Определение двуокиси азота. Гиг. и сан., 1949, 11.
Глава V
СОЕДИНЕНИЯ МЫШЬЯКА, ФОСФОРА, СЕЛЕНА И ТЕЛЛУРА
МЫШЬЯК, МЫШЬЯКОВИСТЫЙ АНГИДРИД И ДРУГИЕ СОЕДИНЕНИЯ МЫШЬЯКА
Мышьяк встречается в нескольких модификациях.
Обычная форма — металлический мышьяк, это кристаллическая масса с металлическим блеском.
Пары мышьяка бесцветны, при резком охлаждении их получается желтый мышьяк, состоящий из прозрачных мягких кубических кристаллов, которые при слабом нагревании, а также под влиянием света переходят в серый мышьяк. Переходным промежуточным продуктом при переходе желтой модификации в металлическую является третья форма мышьяка — черный мышьяк. Мышьяк не растворим в воде.
При нагревании на воздухе мышьяк горит синеватым пламенем, образуя мышьяковистый ангидрид. При горении появляется характерный чесночный запах.
Мышьяковистый ангидрид, называется также белым мышьяком, — белый порошок, растворим в воде, сладковатый раствор его с неприятным металлическим привкусом, имеет слабокислую реакцию. Растворим в щелочах, образует соли.
Соединения мышьяка ядовиты, они поражают нервную систему, органы пищеварения и др.
Предельно допустимая среднесуточная концентрация на мышьяк (неорганические соединения, кроме мышьяковистого водорода в пересчете на мышьяк) 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ МЫШЬЯКА
С ДИЭТИЛДИТИОКАРБАМАТОМ СЕРЕБРА [5]
Принцип определения
Соединения мышьяка восстанавливают цинком в солянокислой среде до мышьяковистого водорода, при взаимодействии последнего с раствором диэтилдитиокарбамата серебра в пиридине, раствор окрашивается в красно-фиолетовый цвет.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Соединения сурьмы мешают определению. Влияние сероводорода устраняют улавливанием его ватой, пропитанной ацетатом свинца.
Аппаратура
1. Прибор для выделения из пробы мышьяковистого водорода. Он состоит из колбы емкостью 50 мл. Колба имеет пришлифованную пробку, в которую впаяны две трубки: верхний конец одной трубки имеет форму воронки, нижний конец трубки доходит до дна. Другая трубка служит для отвода из колбы мышьяковистого водорода.
2. Стеклянная трубка длиной 8 — 9 см наполнена ватой, пропитанной раствором ацетата свинца.
Реактивы
1. Стандартные растворы мышьяка. Исходный раствор с содержанием 100 мкг/мл мышьяка получают растворением в мерной колбе емкостью 100 мл 0,0132 г мышьяковистого ангидрида (сублимированного), 0,1 мл 10% раствора едкого натра и добавляют раствор водой до метки.
Рабочий стандартный раствор с содержанием 2 мкг/мл мышьяка получают разбавлением исходного раствора в 50 раз.
2. Серная кислота, х.ч., не содержащая мышьяка.
3. Азотная кислота, х.ч., 65% раствор.
4. Оксалат аммония, насыщенный раствор.
5. Соляная кислота, 36% раствор, свободная от мышьяка.
6. Йодид калия, х.ч., 1% раствор.
7. Хлорид олова, х.ч., 40% раствор, в соляной кислоте 36% раствор.
8. Цинк зерненный, х.ч., не содержащий мышьяка. Его активируют, обрабатывая разбавленной соляной кислотой.
9. Диэтилдитиокарбамат серебра. 2,25 г диэтилдитиокарбамата натрия растворяют в 100 мл воды и смешивают со 100 мл 1,75% раствора нитрата серебра. Желтый осадок декантируют, отсасывают, промывают водой и высушивают в вакуумном эксикаторе.
Мешает избыток раствора нитрата серебра.
10. Диэтилдитиокарбамат серебра (0,5% раствор в пиридине): 1 г соли растворяют в 200 мл свежеперегнанного пиридина (температура кипения 114 — 116 °C), отфильтрованный раствор сохраняют в темной склянке. Раствор устойчив в течение 2 — 3 мес.
11. Вата, пропитанная ацетатом свинца, для улавливания сероводорода.
В 50 мл воды растворяют 10 г ацетата свинца х.ч., подкисленной уксусной кислотой, прибавляют 20 г глицерина и раствор доводят до 100 мл водой. Вату хлопчатобумажную пропитывают раствором, отжимают избыток и сушат при невысокой температуре. Сохраняют в плотно закрытой банке.
Применяемые реактивы проверяют на отсутствие мышьяка.
Отбор пробы
Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают непрерывно через фильтр АФА в течение суток. Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в один и тот же фильтр.
Ход анализа
Анализ пробы состоит из трех стадий:
1) минерализация пробы;
2) выделение мышьяковистого водорода;
3) фотометрическое определение мышьяка.
В колбу Кьельдаля емкостью 25 мл помещают фильтр с пробой и по капле смачивают всю поверхность фильтра 1 мл серной кислоты (уд. вес 1,84) и 2 мл 65% раствора азотной кислоты. Колбу медленно нагревают на песчаной бане. Когда фильтр растворится, температуру нагревания повышают. Как только раствор в колбе начнет темнеть, прибавляют азотную кислоту по каплям и продолжают нагревать до выделения белых паров серного ангидрида.
Прозрачный раствор в колбе по охлаждении осторожно разбавляют 3 мл раствора оксалата аммония и раствор снова нагревают до выделения белых паров серного ангидрида. Таким образом устраняют остатки окислов азота, связанных в виде нитрозилсерной кислоты. По охлаждении бесцветный и прозрачный раствор из колбы переводят в мерную колбу емкостью 109 мл или в пробирку с притертой пробкой и меткой — 10 мл. Раствор доводят до метки водой.
Для восстановления соединений мышьяка до мышьяковистого водорода 10 мл раствора из колбы Кьельдаля переводят в реакционную колбу, разбавляют его 10 мл воды, прибавляют постепенно 5 мл соляной кислоты, 2 мл раствора йодида калия и 0,5 мл раствора хлорида олова. Колбу закрывают пробкой и конец изогнутой трубки соединяют с трубкой, наполненной ватой, пропитанной ацетатом свинца. К этой трубке присоединяют поглотительный прибор с пористой пластинкой N 1 с 4 мл пиридинового раствора диэтилдитиокарбамата серебра.
Далее в колбу помещают около 3 г цинка, быстро закрывают, помещают в сосуд с водой (если необходимо подогревание раствора).
Выделение водорода должно быть равномерное: если оно замедляется, то прибавляют через воронку оставшуюся часть соляной кислоты.
Во время восстановительного процесса прибор защищают от солнечных лучей. Процесс восстановления протекает 50 — 60 мин., за это время раствор в поглотительном приборе окрашивается, интенсивность окраски измеряют сразу после окончания восстановления в кювете шириной 2 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 533 нм.
Содержание вещества в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 23).
Таблица 23
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
|
Вода, мл |
20 |
19 |
18 |
17 |
16 |
15 |
|
Содержание мышьяка, мкг |
0 |
2 |
4 |
0 |
8 |
10 |
Растворы в реакционных колбах обрабатывают аналогично пробе.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ МЫШЬЯКОВИСТОГО АНГИДРИДА
И ДРУГИХ СОЕДИНЕНИЙ ТРЕХВАЛЕНТНОГО МЫШЬЯКА [3, 8]
Принцип определения
При окислении мышьяковистого ангидрида и других соединений трехвалентного мышьяка смесью перекиси водорода и аммиака образуются соединения пятивалентного мышьяка, которые определяют по синему мышьяково-молибденовому комплексу.
Чувствительность определения 1 мкг в определяемом объеме раствора. Соединения пятивалентного мышьяка, кремния и фосфора мешают определению.
Реактивы
1. Арсенат натрия (трехзамещенный, Na3AsO4 + 2H2O). Исходный стандартный раствор с содержанием 100 мкг мышьяка в 1 мл готовят в мерной колбе емкостью 1 л растворением 0,5661 г соли в воде. Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного.
2. Молибдат аммония (1% раствор в 5 н. растворе серной кислоты).
3. Гидразин сернокислый (0,15% водный раствор, свежеприготовленный).
4. Аммиак (12% раствор).
5. Перекись водорода (10% раствор).
6. Серная кислота (5 н. раствор).
Отбор пробы
Воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, помещенный в патрон. Отбирают около 1000 л исследуемого воздуха.
Ход анализа
Фильтр помещают в стакан, заливают 8 мл аммиака, добавляют 1 мл раствора перекиси водорода и оставляют на 10 мин., периодически помешивая стеклянной палочкой. Далее раствор переносят в фарфоровую чашку, фильтр промывают раствором аммиака. Смывы объединяют с пробой, выпаривают досуха и сухой остаток растворяют в 2 мл дистиллированной воды. Раствор фильтруют через маленький фильтр в мерную пробирку.
Раствор доводят до объема 5 мл. К пробе добавляют 0,5 мл молибдата аммония, 0,2 мл раствора сернокислого гидразина. Смесь взбалтывают и нагревают на водяной бане в течение 10 — 15 мин.
После охлаждения измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см относительно контроля при длине волны 840 нм. Содержание мышьяка в определяемом объеме устанавливают по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов.
На фильтры, употребляемые при отборе проб, наносят различные количества стандартного раствора. Одновременно готовят контроль (табл. 24).
Таблица 24
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Содержание мышьяка, мкг |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
Фильтры обрабатывают аналогично пробе, измеряют величины оптических плотностей и строят график.
Шкалой стандартов можно пользоваться также при визуальном определении, ее готовят одновременно с пробой.
Расчет см. выше.
ФОСФОРНЫЙ АНГИДРИД
Кристаллическое или аморфное вещество белого цвета, плотность 2,39, температура плавления 569°, возгоняется при 359 °C. Сильно гигроскопично. Соединяясь с водой, образует фосфорную кислоту. Растворим в серной кислоте.
Фосфорный ангидрид обладает раздражающим и прижигающим действием на слизистые оболочки и кожу.
Предельно допустимые концентрации: максимальная разовая 0,15 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ФОСФОРНОГО АНГИДРИДА
С МОЛИБДАТОМ АММОНИЯ [6]
Принцип определения
При взаимодействии фосфорной кислоты с молибдатом аммония образуется фосфорно-молибденовый комплекс, который восстанавливают до фосфорно-молибденовой сини.
Чувствительность определения 0,20 мкг в анализируемом объеме раствора.
Соли мышьяка мешают определению.
Реактивы
1. Стандартный раствор фосфорного ангидрида. Исходный раствор с содержанием 100 мкг/мл фосфорного ангидрида готовят растворением в воде 0,0219 г однозамещенного фосфата натрия или 0,0504 г двузамещенного фосфата натрия в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного в 100 раз.
Отбор проб, обработку фильтров и приготовление реактивов проводят так же, как при определении мышьяковистого ангидрида по реакции с молибдатом аммония, но без окисления (см. выше).
КАРБОФОС
|
C10H19O6PS2 |
Мол. вес 330,37 |
Маслянистая бесцветная жидкость со слабым неприятным запахом, не растворима в воде, но хорошо растворима в органических растворителях.
Продажный карбофос является 35% концентратом — это густая жидкость от светло-желтого до темно-коричневого цвета с сильным неприятным запахом.
Карбофос — токсичное вещество.
Предельно допустимая концентрация: максимальная разовая — 0,015 мг/м3.
ОПРЕДЕЛЕНИЕ КАРБОФОСА С МОЛИБДАТОМ АММОНИЯ [7]
Принцип определения
При взаимодействии карбофоса со смесью кислот азотной и серной в присутствии перманганата калия происходит разложение карбофоса и выделение фосфорной кислоты, которая с молибдатом аммония образует комплекс. Последний при восстановлении превращается в молибденовую синь, по которой определяют содержание карбофоса.
Чувствительность определения 2,13 мкг карбофоса в анализируемом объеме раствора.
Фосфаты и другие фосфорноорганические соединения мешают определению.
Реактивы
1. Поглотительный раствор: уксусная кислота и вода в отношении 4:1.
2. Стандартный раствор фосфора: исходный раствор с содержанием 100 мкг/мл получают растворением 0,1159 г двузамещенного фосфата натрия подкисленной водой (2 — 3 капли кислоты в мерной колбе емкостью 100 мл).
Рабочие стандартные растворы с содержанием 10 мкг/мл (А) и 1 мкг/мл (Б) получают разбавлением исходного раствора в 10 и 100 раз.
3. Молибдат аммония (0,25% раствор: 0,25 г соли растворяют в 100 мл 1,25 н. раствора серной кислоты).
4. Окислительная смесь: 0,5 г перманганата калия хорошо растирают и растворяют в 10 мл серной кислоты, уд. вес 1,84 (срок годности смеси 5 дней).
5. Восстановительный раствор: 1 г хлорида олова растворяют в 10 мл соляной кислоты, уд. вес 119. Затем 0,1 мл раствора доводят бутанолом до 20 мл. Раствор годен один день.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористыми фильтрами N 1, содержащие по 5 мл поглотительного раствора в каждом. Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора отдельно переносят 4 мл пробы в фарфоровый тигель и вносят 2,5 мл азотной кислоты и 0,1 мл окислительной смеси.
Тигли помещают на слегка подогретую песчаную баню и температуру бани медленно доводят до 80 — 90°. Раствор нагревают до прекращения выделения уксусной кислоты и окислов азота. Затем поднимают температуру бани до 180° (не выше) и раствор выпаривают досуха. По охлаждении остаток растворяют в 1 — 1,5 мл воды. Растворение проводят путем добавления воды по 0,5 мл 2 — 3 раза, переводя раствор в пробирку. Затем наливают 0,3 мл молибдата аммония и через 15 мин. вносят 0,4 мл бутанола. Пробирку плотно закрывают пробкой и содержимое пробирки встряхивают (40 — 50 раз), проводят экстракцию. После разделения слоев отбирают 0,3 мл бутанолового раствора и переносят его в сухую пробирку, в которую вливают 0,9 мл раствора восстановителя.
Через 10 мин. содержание вещества определяют по шкале стандартов, которую готовят одновременно с пробами (табл. 25).
Таблица 25
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл (Б) |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
— |
— |
|
Рабочий стандартный раствор, мл (А) |
— |
— |
— |
— |
— |
0,1 |
0,15 |
|
Вода, мл |
1 |
0,8 |
0,7 |
0,6 |
0,5 |
0,9 |
0,85 |
|
Содержание фосфора, мкг |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
1,0 |
1,15 |
|
Содержание карбофоса <1>, мкг |
0 |
2,13 |
3,18 |
4,26 |
5,30 |
10,6 |
12,19 |
———————————
<1> Коэффициент пересчета с фосфора на карбофос 10,67.
Все пробирки шкалы обрабатывают аналогично пробе. Расчет см. выше.
БУТИФОС
Жидкость от желтого до темно-коричневого цвета, температура кипения 154 — 174°. Нерастворима в воде, образует с водой эмульсию, растворима в органических растворителях; обладает сильным неприятным запахом. Раздражает слизистые оболочки носа и глаз.
Предельно допустимые концентрации: максимальная разовая — 0,01 мг/м3, среднесуточная — 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ БУТИФОСА С МОЛИБДАТОМ АММОНИЯ [4]
Метод основан на разложении бутифоса смесью азотной и серной кислот в присутствии перманганата калия и определении выделяющейся фосфорной кислоты с молибдатом аммония.
Чувствительность определения 2 мкг бутифоса в анализируемом объеме раствора.
Реактивы, отбор проб и проведение анализа аналогичны карбофосу (см. выше).
Коэффициент пересчета с фосфора на бутифос 10,1.
Препарат М-81 [7]
(0,0-диметил-![]() -этилмеркаптоэтилдитиофосфат)
-этилмеркаптоэтилдитиофосфат)
|
(CH3O)2PSSCH2CH2SC2H5 |
Мол. вес 246,36 |
Препарат М-81 — бурая жидкость с неприятным запахом, легко смешивающаяся с водой с образованием эмульсии. Применяется как инсектицид, выпускается в виде 50% концентрата в смеси с ОП-7. М-81 обладает высокой токсичностью. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,001 мг/м3.
Определение препарата М-81 см. «Определение карбофоса» (см. выше).
СЕЛЕН
Селен может быть в аморфном и кристаллическом состоянии. Температура плавления аморфного селена 50°, кристаллического 220°, плотность 4,26 и 4,2 соответственно. Аморфный селен — бурая масса, кристаллический — вещество с металлическим блеском. Селен растворим в воде, в крепком растворе сульфита натрия, в смеси соляной кислоты с бромом, в крепкой серной кислоте. Азотная кислота окисляет селен до селенистого ангидрида. Металлический селен менее ядовит, чем его соединения. Характерные признаки отравления — раздражение слизистых оболочек, общая слабость, головная боль. Предельно допустимая концентрация не установлена.
ОПРЕДЕЛЕНИЕ СЕЛЕНА С 3,31-ДИАМИНОБЕНЗИДИНОМ [1]
Принцип определения
При взаимодействии селена с 3,31-диаминобензидином образуется комплексное соединение, окрашенное в желтый цвет. Чувствительность определения 0,5 мкг в анализируемом объеме раствора. Теллур, медь, железо, мышьяк не мешают определению.
Реактивы
1. Стандартный раствор селена. Исходный раствор селена готовят растворением 500 мг селена при умеренном нагревании на водяной бане в 10 мл соляной кислоты с добавлением 5 — 7 капель азотной кислоты: раствор переводят в мерную колбу емкостью 500 мл и доводят до метки водой. 1 мл раствора содержит 1 мг селена. Рабочий стандартный раствор с содержанием 1 мл/1 мкг селена готовят разбавлением исходного раствора в 1000 раз. Раствор должен быть свежеприготовленным.
2. Соляная кислота, удельный вес 1,19.
3. Азотная кислота, концентрированная.
4. Смесь соляной и азотной кислот, взятых в соотношении 1:1.
5. Комплексон III (динатриевая соль этилендиаминотетрауксусной кислоты) (2,5% раствор).
6. Кислота муравьиная (разбавленная 1:9).
7. Аммиак (разбавленный 1:1).
8. Крезоловый красный (0,1% раствор 100 мг индикатора растворяют в 20 мл 1% раствора едкого натра и доводят до 100 мл).
9. Толуол.
10. 3,31-Диаминобензидин (0,5% раствор). Раствор должен быть свежеприготовленным.
Отбор пробы
Воздух со скоростью 20 л/мин. протягивают через фильтр АФА-В-18, помещенный в патрон. Для анализа следует отобрать 500 л воздуха.
Ход анализа
Фильтр с пробой переносят в стакан и обрабатывают 10 мл смесью соляной и азотной кислот при умеренном нагревании (избегать бурной реакции, которая может привести к улетучиванию селена). Полученный раствор упаривают почти досуха, и нагревание сухого остатка допускать нельзя ввиду возможного улетучивания селена. К осадку добавляют 10 — 15 мл горячей воды и содержимое нагревают до получения прозрачного раствора. К раствору добавляют 2 — 3 капли крезолового красного, 1 мл комплексона III и 2 мл муравьиной кислоты, тщательно перемешивают и нейтрализуют аммиаком до желтой окраски (pH 2,0 — 3,0). К холодному раствору приливают 2 мл свежеприготовленного раствора 3,31-диаминобензидина и ставят в темноту на 30 мин. По истечении времени раствор нейтрализуют аммиаком до появления фиолетовой окраски (pH 8,0), переводят в делительную воронку емкостью 50 — 75 мл и добавляют 5 мл толуола. Воронку энергично встряхивают в течение минуты и после разделения слоев водную фазу отбрасывают, а слой толуола через сухой фильтр сливают в кювету с толщиной слоя 1 см и фотометрируют при длине волны 420 нм по сравнению с контролем. Содержание селена определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, которые употребляют при отборе проб. Одновременно готовят контрольный фильтр (табл. 26).
Таблица 26
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий, стандартный раствор, мл |
0 |
0,5 |
0,7 |
1,0 |
1,2 |
1,5 |
1,7 |
2,0 |
|
Содержание селена, мкг |
0 |
0,5 |
0,7 |
1,0 |
1,2 |
1,5 |
1,7 |
2,0 |
Контрольный фильтр и фильтры, содержащие различное количество селена, обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках.
Расчет см. выше.
ТЕЛЛУР
Вещество серебристо-белого цвета. Температура плавления 452 °C, температура кипения 1390 °C. Сгорает с образованием двуокиси теллура. В воде не растворим, не растворим в кислотах, не являющихся окислителями. Растворим в концентрированной серной, азотной кислотах, растворим в царской водке и концентрированных растворах щелочей. Теллур и его соединения токсичны.
Действие их сходно с действием неорганических соединений мышьяка и серы. При попадании теллура в организм через органы дыхания образуется летучее соединение — теллуристый метил, обусловливающий появление специфического чесночного запаха в выдыхаемом воздухе.
ОПРЕДЕЛЕНИЕ ТЕЛЛУРА И ЕГО СОЕДИНЕНИЙ С БУТИЛРОДАМИНОМ С [2]
Принцип определения
При взаимодействии теллура с бутилродамином С образуется окрашенный комплекс, который экстрагируется бензолом.
Чувствительность определения 0,5 мкг теллура в определяемом объеме раствора. Определению мешает медь. Селен определению не мешает.
Реактивы
1. Исходный стандартный раствор с содержанием 1 мг/мл готовят, помещая 0,1 г теллура в выпарительную чашку, в которую добавляют 10 мл концентрированной серной кислоты. Содержимое выпаривают досуха, после охлаждения добавляют 20 мл концентрированной соляной кислоты и переносят в мерную колбу емкостью 100 мл и доводят объем ее до метки водой.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят соответствующим разбавлением исходного.
2. Серная кислота, уд. вес 1,84, разведенная 1:1.
3. Азотная кислота, уд. вес 1,4.
4. Соляная кислота, уд. вес 1,19; 0,1 и 0,5 н. растворы.
5. Трибутилфосфат, 30% раствор в ксилоле (по объему).
6. Ацетон.
7. Бензол.
8. Бромид натрия (2 М раствор).
9. Бутилродамин С (0,1% водный раствор).
10. Аскорбиновая кислота (2% водный раствор, свежеприготовленный).
Отбор пробы
Воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, помещенный в патрон. Для анализа отбирают не менее 600 — 700 л исследуемого воздуха.
Ход анализа
Фильтр помещают в стакан емкостью 50 мл и заливают смесью концентрированных серной и азотной кислот (1,5:1). После полного растворения фильтра смесь упаривают почти досуха. Влажный остаток обрабатывают 5 мл 5 н. раствором соляной кислоты. Содержимое стакана переносят в делительную воронку емкостью 25 мл. Добавляют 5 мл трибутилфосфата в ксилоле и взбалтывают в течение 2 мин. После разделения к органической фазе добавляют 5 мл 0,1 н. раствора соляной кислоты и реэкстрагируют теллур в течение 2 мин. Полученный реэкстракт осторожно упаривают, не допуская обугливания органики. Остаток обрабатывают 5,5 мл раствора серной кислоты 1:1, добавляют 0,5 мл 2 М раствора бромида натрия, 1 мл 0,1% раствора бутилродамина С и 2 мл раствора аскорбиновой кислоты.
В охлажденную смесь вносят 1 мл ацетона.
Смесь переносят в делительную воронку, в которую вносят 5 мл бензола, и экстрагируют в течение 1 мин. Оптическую плотность бензольного раствора измеряют в кюветах с толщиной слоя 1 см при длине волны 560 нм относительно контроля.
Количество теллура устанавливают по калибровочному графику. Для его построения готовят шкалу стандартов, при этом наносят на фильтры стандартный раствор (табл. 27).
Таблица 27
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
|
Содержание теллура, мкг |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
Фильтры с нанесенными стандартными растворами и контроль обрабатывают аналогично пробе. По полученным значениям оптических плотностей строят калибровочный график. Шкалой можно пользоваться при визуальном определении, ее готовят одновременно с пробой.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева Л.С. Спектрофотометрическое определение селена в атмосферном воздухе. Гиг. и сан., 1970, 2.
2. Алексеева Л.С., Мурашова В.И. и др. Фотометрическое определение теллура в атмосферном воздухе. Заводская лаборатория, т. XXXVIII, 11, 1971.
3. Гольдберг Е.Х. Определение мышьяковистого ангидрида и других неорганических соединений мышьяка. Материалы Юбилейной конференции по общей и коммунальной гигиене. М., Санитарно-эпидемиологическая станция г. Москвы, 1967.
4. Мухамедова С.Ю. Экспериментальные данные к нормированию бутифоса в атмосферном воздухе. Гиг. и сан., 1968, 12.
5. Определение соединений мышьяка. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. Ч. 1. М., 1970.
6. Определение фосфорного ангидрида. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. Ч. II. М., 1973.
7. Технические условия на метод определения карбофоса, метафоса, М-81, октаметила и др. В. III, 1963.
8. Технические условия на метод определения мышьяковистого ангидрида. В. IV, 1965.
Глава VI
МЕТАЛЛЫ
ВАНАДИЙ
Металл серебристо-серого цвета.
Плотность 6,11. Температура плавления 1900 +/- 25 °C.
Температура кипения 3330 °C. Не растворим в воде. Растворим в концентрированных серной, азотной, фтористо-водородной кислотах. Известны окисные соединения: трехокись, пятиокись, ванадат аммония, йодистый ванадий, бромистый ванадий и др.
Соединения ванадия токсичны. Вызывают изменения в органах дыхания, нервной системе, кровообращении, обмене веществ, обладают раздражающим действием.
Данных о вредном действии металлического ванадия не имеется.
Предельно допустимая концентрация пятиокиси ванадия: среднесуточная 0,002 мг/м3.
ОПРЕДЕЛЕНИЕ ВАНАДИЯ С САЛИЦИЛГИДРОКСАМОВОЙ КИСЛОТОЙ [1]
Принцип определения
При окислении соединений ванадия образуется пятиокись ванадия, которая с салицилгидроксамовой кислотой дает ванадий-салицилгидроксамовый комплекс, окрашенный в фиолетовый цвет.
Чувствительность определения — 1 мкг ванадия в анализируемом объеме раствора.
Железо, кальций, магний, марганец, титан, медь, кремний, никель, цинк до 500 мкг не мешают определению.
Реактивы
1. Стандартный раствор ванадия готовят из ванадата аммония NH4VO3. Реактив дважды перекристаллизовывают из горячей воды, охлаждают раствор льдом, отделяют маточный раствор, кристаллы промывают этанолом и сушат на воздухе, затем в сушильном шкафу при 50 °C.
Исходный раствор с содержанием 1 мг/мл готовят растворением 0,2295 г перекристаллизованного ванадата аммония в мерной колбе емкостью 100 мл. Навеску помещают в колбу, растворяют в 50 мл горячей воды (70 — 75°). После растворения и охлаждения раствор доводят до метки.
Разбавляя исходный раствор в 100 раз, получают рабочий стандартный раствор с содержанием 10 мкг/мл.
2. Этиловый спирт.
3. Азотная кислота, уд. вес 1,4, 50% раствор (по объему).
4. Натр едкий, х.ч., 5% раствор; 8,5 и 20% раствор.
5. Уксусная кислота, ледяная.
6. Метиловый эфир салициловой кислоты.
7. Гидроксиламин солянокислый. 35 г гидроксиламина растворяют в 100 мл 20% раствора едкого натра. Хранят в холодильнике или применяют свежеприготовленным.
8. Соляная кислота, уд. вес 1,19.
9. Салицилгидроксамовая кислота, 0,5% раствор в ледяной уксусной кислоте.
Синтез салицилгидроксамовой кислоты: 76 г метилового эфира салициловой кислоты помещают в 300 мл охлажденного 8,5% раствора едкого натра, добавляют 900 мл воды и перемешивают раствор в течение 45 — 50 мин. Затем вносят 100 мл охлажденного раствора гидроксиламина и оставляют на 24 ч. Все операции проводят при температуре не выше 25°. Далее раствор фильтруют, устанавливают величину pH, равную 1 — 2 (по универсальной индикаторной бумаге). Осадок отделяют при помощи воронки Бюхнера, промывают водой, подкисленной соляной кислотой. Выход салицилгидроксамовой кислоты составляет 70 — 72%, температура плавления 140 — 145 °C. Реактив устойчив в течение нескольких лет.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью 50 — 100 л/мин. протягивают через фильтр АФА, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 10 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток. Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз со скоростью 5 — 10 л/мин. с перерывом в 4 или 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, переносят в фарфоровый тигель, добавляют 2 мл 50% азотной кислоты, выпаривают досуха на водяной бане или электроплитке, закрытой асбестом. Затем помещают в муфельную печь и постепенно повышают температуру до 500 °C. После полного озоления пробы тигель охлаждают, добавляют 2 мл 5% раствора едкого натра и 5 мл воды. Нагревают тигель в течение 2 — 3 мин. на кипящей водяной бане и содержимое фильтруют в пробирку через маленький фильтр; тигель несколько раз обмывают дистиллированной водой и доводят объем пробы до 10 мл. Берут 5 мл пробы, вносят 5 мл 5% раствора салицилгидроксамовой кислоты, перемешивают. Через 15 — 20 мин. интенсивность окраски измеряют в кювете с толщиной слоя 2 см при длине волны 490 нм по сравнению с контролем. Содержание ванадия во всей пробе определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры АФА (табл. 28). Одновременно готовят контрольный фильтр.
Таблица 28
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание ванадия, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Контрольный фильтр и фильтры, содержащие различные количества ванадия, помещают в тигли и обрабатывают аналогично пробам.
Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках одновременно с пробой.
Коэффициенты пересчета ванадия на:
1. Ванадия окись VO 1,31.
2. Ванадия трехокись V2O3 1,47.
3. Ванадия двуокись VO2 1,62.
4. Ванадия пятиокись V2O5 1,78.
5. Ванадия карбид VC 1,23.
6. Ванадат аммония NH4VO3 2,3.
7. Ванадат калия (мета-) KVO3 2,7. 3
8. Ванадат натрия (мета-) NaVO3 2,39.
Ванадат натрия (орто-) NO2VO4 3,61.
9. Феррованадий FeV 2,23.
Расчет см. выше.
Примечание. При определении только пятиокиси ванадия исключают окисление и фильтры не обрабатывают азотной кислотой.
ХРОМ
Металл серебристо-серого цвета. Плотность 7,16. Температура плавления 1875 °C, температура кипения 2480 °C.
Не растворим в холодной и горячей воде и азотной кислоте, растворим в соляной и серной кислотах.
Соединения хрома могут иметь валентность от двух до шести. В хроматах он трехвалентен, в бихроматах — шестивалентен.
Известны окисные соединения хрома — окись хрома CrO3 зеленого цвета, хромовый ангидрид — желтого цвета, соли трех- и шестивалентного хрома.
Соли шестивалентного хрома легко восстанавливаются в кислой среде до трехвалентного состояния.
Соединения хрома токсичны, особенно ядовиты шестивалентные соединения, меньшей токсичностью характеризуются трехвалентные соединения. Хром и его двухвалентные соединения малотоксичны.
Соединения хрома обладают раздражающим и прижигающим действием, они оказывают также общетоксическое действие. При контакте с хромом отмечена повышенная заболеваемость раком дыхательных путей.
Предельно допустимые концентрации хрома шестивалентного в пересчете на CrO3: максимальная разовая 0,0015 мг/м3, среднесуточная 0,0015 мг/м3.
ОПРЕДЕЛЕНИЕ ХРОМОВОГО АНГИДРИДА И СОЛЕЙ ХРОМОВОЙ
КИСЛОТЫ (Cr6) С ДИФЕНИЛКАРБАЗИДОМ [3]
Принцип определения
При взаимодействии шестивалентного хрома с дифенилкарбазидом в кислой среде происходит образование соединения, окрашенного в красно-фиолетовый цвет.
Чувствительность определения — 0,4 мкг хрома в анализируемом объеме.
Железо, ванадий, молибден мешают определению в количестве более 1 мг.
Реактивы
1. Стандартный раствор хрома. Исходный раствор с содержанием 100 мкг/мл хрома готовят растворением 0,0282 г перекристаллизованного бихромата калия в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением разбавлением исходного в 100 раз.
2. Соляная кислота, удельный вес 1,4.
3. Этанол.
4. Смесь 1 мл соляной кислоты и 100 мл этанола.
5. Дифенилкарбазид. 0,5 г дифенилкарбазида растворяют во всем объеме смеси соляной кислоты с эталоном.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, беззольный фильтр, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 10 — 15 мин. протягивают через фильтр, укрепленный в патроне, в течение суток непрерывно. Допустимо отбор проб проводить 6 — 12 раз в один и тот же фильтр с перерывами в 2 или 4 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в стакан и заливают 5 мл горячей воды. Помешивают стеклянной палочкой. Через 10 — 15 мин. 4 мл прозрачного раствора переносят в пробирку, вносят 0,4 мл раствора дифенилкарбазида, перемешивают. Через 10 — 15 мин. интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя в 1 или 2 см при длине волны 540 нм по сравнению с контролем. Содержание хрома в анализируемом объеме (4 мл) определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов. При этом рабочий стандартный раствор наносят на фильтры, употребляемые для отбора проб (табл. 29). Одновременно готовят и контрольный фильтр.
Таблица 29
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
|
Содержание хрома, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
Коэффициент пересчета с хрома на CrO3 1,94.
Контрольный фильтр и фильтры, содержащие различные количества хрома, помещают в стаканы, обрабатывают аналогично пробе.
Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках одновременно с пробами, как указано в табл. 29.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СОЕДИНЕНИЙ ДВУХ- И ТРЕХВАЛЕНТНОГО ХРОМА [5]
Принцип определения
Соединения двух- и трехвалентного хрома окисляют перманганатом калия. Образующиеся соединения шестивалентного хрома при взаимодействии с дифенилкарбазидом дают окрашенные в розово-фиолетовый цвет соединения.
Чувствительность определения — 1 мкг шестивалентного хрома в анализируемом объеме раствора. Определению мешают железо, ванадий, медь, молибден, ртуть в количествах, во много раз превышающих содержание хрома.
Реактивы
1. Азотная кислота (1,4), разбавленная 1:1, 2 н. раствор.
2. Серная кислота (1,84), 0,5 н. раствор.
3. Перманганат калия, 0,2 н. раствор.
4. Фосфорная кислота, концентрированная.
5. Этиловый спирт, 96°.
6. Аммиак, 25% раствор.
7. Дифенилкарбазид, 0,5% раствор. Готовят в мерной колбе емкостью 100 мл, в которую вносят 0,5 г дифенилкарбазида, 1 мл концентрированной соляной кислоты, доводят до метки этанолом.
Приготовление стандартного раствора и отбор пробы см. «Определение шестивалентного хрома».
Ход анализа
Фильтр заливают 40 мл 2 н. раствором азотной кислоты и нагревают на песчаной бане в течение 15 мин. Раствор фильтруют, промывают фильтр 2 н. раствором азотной кислоты и водой. Фильтрат выпаривают и пробу помещают в муфельную печь. Температура печи не должна превышать 550°. Сжигание проводят в течение 30 мин. Далее пробу растворяют в 7 мл 0,5 н. раствора серной кислоты и подогревают на водяной бане. Нерастворимый остаток отфильтровывают. Ставят пробу на водяную баню, нагретую до 100 °C, и добавляют по каплям 0,2 н. раствора перманганата калия до получения устойчивого розового окрашивания. Нагревают в течение 15 мин. и вводят по каплям этиловый спирт до обесцвечивания. Пробу переносят в мерную колбу емкостью 25 мл, добавляют 1 мл фосфорной кислоты и дистиллированной воды до объема около 20 мл. Затем добавляют 1 мл раствора дифенилкарбазида и доводят объем колбы до метки. Измеряют оптическую плотность в кюветах с шириной слоя 1 см при длине волны 540 нм. Калибровочный график строят на основании приготовленной шкалы стандартов. Для этого на фильтры, употребляемые при отборе, наносят стандартный раствор. Фильтры обрабатывают аналогично пробе, одновременно готовят контрольный фильтр.
Шкалу строят с содержанием от 1 до 30 мкг хрома.
Примечание. Влияние железа устраняют добавлением фосфорной кислоты. Влияние остальных металлов можно устранить экстрагированием хлороформом, за исключением ртути, которую связывают добавлением раствора хлористого натрия. В случае присутствия значительных количеств железа (более 1000 мкг) его осаждают. Для этой цели после добавления этанола для восстановления перманганата калия в пробу вводят раствор аммиака до щелочной реакции. При подогревании образуется гидроокись железа, которую удаляют фильтрованием. Аммиак удаляют подогреванием до получения нейтральной реакции. Далее анализ проводят, как указано выше.
Расчет см. выше.
МАРГАНЕЦ
Металл серовато-серебристого цвета. Окисная пленка сообщает металлу розовую окраску. Плотность 7,2.
Температура плавления 1244 °C, температура кипения 2120 °C.
Растворим в уксусной и минеральных кислотах. Соединения марганца могут иметь валентность от 2+ до 7+.
Известны окисные соединения марганца. Закись — MnO, окись — Mn3O4 , двуокись — MnO2, марганцовистый ангидрид — Mn2O7. Водные растворы марганцовистой кислоты окрашены в зеленый цвет, марганцовой — в фиолетовый.
Соединения марганца токсичны, действуют на центральную нервную систему, легкие, печень, вызывают изменения состава крови.
Предельно допустимая концентрация марганца и его соединений в пересчете на MnO2: среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ МАРГАНЦА С ПЕРСУЛЬФАТОМ АММОНИЯ [8]
Принцип определения
При взаимодействии иона марганца с персульфатом аммония в присутствии нитрата серебра, играющего роль катализатора, происходит окисление марганца с образованием марганцовой кислоты, окрашенной в фиолетовый цвет.
Чувствительность определения — 2 мкг в определяемом объеме раствора.
Хром мешает определению.
Реактивы
1. Стандартный раствор марганца: исходный раствор с содержанием 100 мкг/мл марганца готовят растворением 0,0504 г сульфата марганца MnSO4·7H2O в 5% растворе серной кислоты в мерной колбе емкостью 100 мл.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят разбавлением исходного в 100 раз.
2. Серная кислота, уд. вес 1,82 — 1,84, 5% раствор.
3. Щавелевая кислота, 8% раствор.
4. Смесь кислот серной (уд. вес 1,82 — 1,84) и 8% щавелевой в отношении 1:1 готовят перед употреблением.
5. Персульфат аммония, кристаллический.
6. Нитрат серебра, 0,1% раствор.
7. Ортофосфорная кислота, 20% раствор.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 50 л/мин. протягивают через фильтр АФА, беззольный, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации исследуемый воздух со скоростью до 10 л/мин. протягивают через фильтр, укрепленный в патроне, в течение суток непрерывно.
Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывами в 4 или в 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в фарфоровую чашку. Осторожно озоляют при 500 — 600 °C. К озоленной пробе добавляют 0,5 мл смеси кислот, помещают чашку на песчаную баню и нагревают до удаления пузырьков углекислоты и белых паров серного ангидрида.
Образовавшийся сульфат марганца растворяют в 1 мл 5% раствора серной кислоты. Раствор фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в который вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см. Чашку тщательно обмывают несколькими порциями 5% раствора серной кислоты, растворы фильтруют.
Общий объем пробы доводят до 4 мл. Для анализа берут до 3,5 мл пробы. Можно в чашку сразу добавить 4 мл 5% серной кислоты, перенести раствор в центрифужные пробирки и центрифугировать, и в этом случае для анализа берут до 3,5 мл прозрачного раствора. Вносят 0,1 мл нитрата серебра, по 0,1 мл ортофосфорной кислоты и добавляют несколько кристалликов персульфата аммония; перемешивают и пробирки с пробами помещают на водяную баню, нагретую до 70 °C на 5 мин. После охлаждения интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя 1 или 2 см при длине волны 545 пм по сравнению с контролем. По предварительно построенному калибровочному графику устанавливают содержание марганца в анализируемом объеме (4 мл).
Для построения калибровочного графика готовят шкалу стандартов. При этом рабочий стандартный раствор наносят на такие же фильтры, как и употребляемые для отбора пробы (табл. 30). Одновременно обрабатывают и контрольный фильтр.
Таблица 30
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
2 |
3 |
5 |
7 |
9 |
|
Содержание марганца, мкг |
0 |
2 |
3 |
5 |
7 |
9 |
Контрольный фильтр и фильтры, содержащие различные количества марганца, помещают в фарфоровые чашки, озоляют. Контрольный фильтр и шкалу обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках, одновременно с пробами, так же, как указано в табл. 30.
Коэффициент пересчета с Mn на MnO2 1,58.
Расчет см. выше.
КОБАЛЬТ
Металл серебристо-серого цвета. Плотность 8,8. Температура плавления 1492 °C, температура кипения 2255 °C. Растворим в азотной, соляной кислотах. Серная кислота не действует на кобальт на холоде. Стоек по отношению к воздуху и воде. Кобальт и его соединения токсичны. Действуют на верхние дыхательные пути, почки, вызывают тромбоз сосудов и др.
НИКЕЛЬ
Металл серебристо-белого цвета. Плотность 8,9. Температура кипения 2140 °C, температура плавления 1453 °C. Плохо растворим в воде, растворим в разбавленной азотной, соляной, серной кислотах. Никель относится к мало активным элементам, окисляется лишь при 500 °C. Никель и его соединения токсичны. Аэрозоли их раздражают верхние дыхательные пути, изменяют состав крови.
МЕДЬ
Металл бледно-розового цвета. Плотность 8,92. Температура плавления 1083 °C, температура кипения 2580 °C. Растворима в азотной и концентрированной серной кислотах. Реагирует с соляной кислотой и гидратом окиси аммония. На воздухе окисляется, образуя окисную пленку. Соединения меди токсичны. Действуют на кожные покровы, слизистые оболочки, вызывая высыпания, зуд, головную боль, лихорадку.
ОПРЕДЕЛЕНИЕ МЕДИ, КОБАЛЬТА И НИКЕЛЯ ХРОМАТОГРАФИЕЙ
НА БУМАГЕ [4]
Принцип определения
Медь, кобальт, никель переводятся в растворимые соединения обработкой крепкими кислотами и разделяются на бумаге в системе бутанол — ацетон — соляная кислота — вода.
Для проявления разделенных зон применяют рубеано-водородную кислоту и аммиак. Медь окрашивается в серо-зеленый, кобальт — в желтый, никель — сине-фиолетовый цвета.
Чувствительность определения — 0,1 мкг кобальта и никеля и 0,2 мкг меди на хроматограмме.
Определению не мешают алюминий, цинк, олово, марганец.
Влияние железа устраняется в процессе обработки пробы, свинец маскируется в процессе проявления хроматограммы.
Аппаратура
1. Спектрофотометр СФ-10, СФ-14. Плосковыпуклые линзы с оптической силой +10D.
2. Хроматографическая камера (эксикатор).
Реактивы
1. Соляная кислота концентрированная, х.ч., 7 н. раствор, 2 н. раствор, 0,1 н. раствор, 7 н. раствор, насыщенный диэтиловым эфиром. Последний раствор готовят следующим образом. 7 н. раствор соляной кислоты охлаждают до 10 °C, встряхивают с небольшой порцией эфира и снова охлаждают. Эту операцию повторяют до тех пор, пока над кислотой не останется слой эфира. Смесь готовят перед употреблением.
2. Азотная кислота, концентрированная, х.ч.
3. Эфир диэтиловый (для наркоза).
4. Спирт бутиловый, нормальный, х.ч.
5. Ацетон, х.ч.
6. Спирт этиловый, перегнанный при 78 °C.
7. Серная кислота, концентрированная, х.ч.
8. Рубеановодородная кислота, 0,1% раствор в этиловом спирте.
9. Виннокаменная кислота, 4% раствор в этиловом спирте.
10. Аммиак, 25% раствор.
11. Бумага хроматографическая «средняя».
12. Система растворителей. Спирт бутиловый 76 мл, ацетон 36 мл, соляная кислота концентрированная 44 мл и 14 мл воды вносят в делительную воронку и перемешивают.
13. Стандартный раствор никеля. Исходный раствор готовят растворением 0,4049 г хлорида никеля в 100 мл 7 н. раствора соляной кислоты. 1 мл раствора содержит 1 мг никеля. Рабочий стандартный раствор готовят соответствующим разбавлением 7 н. раствором соляной кислоты. Рабочий стандартный раствор содержит 100 мкг/мл.
14. Стандартный раствор кобальта. Исходный раствор с содержанием кобальта 1 мг/мл готовят растворением 0,4037 г хлорида кобальта в 100 мл 7 н. раствора соляной кислоты. Рабочий стандартный раствор с содержанием 100 мкг кобальта в 1 мл готовят соответствующим разбавлением исходного раствором 7 н. соляной кислоты.
15. Стандартный раствор меди. Исходный раствор с содержанием меди 1 мг/мл готовят растворением 0,2667 г хлорида меди в 100 мл 7 н. раствора соляной кислоты. Рабочий стандартный раствор с содержанием 100 мкг меди в 1 мл готовят соответствующим разбавлением 7 н. раствором соляной кислоты.
16. Смесь стандартных растворов с содержанием по 10 мкг меди, никеля и кобальта в объеме 1 мл. В мерную колбу емкостью 25 мл вносят по 2,5 мл рабочих стандартных растворов меди, кобальта, никеля. Объем колбы доводят до метки 7 н. раствором соляной кислоты.
Отбор пробы
Исследуемый воздух со скоростью 50 л/мин. протягивают через фильтр АФА, укрепленный в патроне, в течение 20 — 30 мин.
Ход анализа
Фильтр переносят в фарфоровый тигель, добавляют 1,5 мл концентрированной соляной кислоты, 0,5 мл азотной кислоты (1,4) и нагревают в течение 5 — 10 мин., не допуская кипения. Затем добавляют 0,25 мл серной кислоты и выпаривают досуха. Сухой остаток помещают в муфельную печь, постепенно доводят температуру до 500°.
После охлаждения зольный остаток растворяют в 1,5 мл соляной кислоты (1,19), вносят 0,5 мл азотной кислоты (1,4 уд. вес) и выпаривают досуха при слабом нагревании.
Остаток обрабатывают 3 мл 7 н. раствора соляной кислоты и переносят в делительную воронку. Тигель обмывают 7 н. раствором соляной кислоты, доводят объем до 5 мл. Затем в делительную воронку вносят 5 мл диэтилового эфира, встряхивают в течение 2 — 3 мин. После разделения слоев нижний сливают в тигель. Верхний слой, эфирный, промывают 1 мл 7 н. раствором соляной кислоты, насыщенным эфиром. Полученный после промывания водный слой сливают в тигель.
Если эфирный слой окрашен в желтый цвет, то водный слой подвергают повторной обработке до тех пор, пока экстрагент не станет бесцветным. Далее водный слой, находящийся в тигле, выпаривают до получения влажного остатка, который обрабатывают 0,5 мл 0,1 н. раствора соляной кислоты.
На листе хроматографической бумаги шириной 95 мм и длиной 220 мм на расстоянии 18 мм от нижнего края во всю ширину намечается линия старта. От нее вверх вырезают полосы шириной 6,5 мм и высотой 80 мм. Расстояние между полосами 10 мм. Для очистки бумаги ее помещают в хроматографическую камеру, на дно которой вносят 2 н. раствор соляной кислоты. После поднятия кислоты на высоту 150 мм бумагу вынимают и высушивают. Хранят в бумажном пакете не более 6 дней.
На бумагу наносят 0,05 мл пробы, обдувая ее воздухом, подогретым теплоэлектровентилятором. Разделение металлов проводят в хроматографической камере, на дно которой за 10 мин. до хроматографирования вносят систему растворителей. Бумагу закладывают так, чтобы нижний край погружался в растворитель на 1 см. Когда растворитель продвинется на высоту 8 см от линии старта, бумагу извлекают, высушивают на воздухе и проявляют раствором виннокаменной кислоты путем двустороннего орошения, а затем через 1 — 2 мин. раствором рубеановодородной кислоты с последующей обработкой парами аммиака в течение 2 — 3 мин. При наличии меди наблюдается серо-зеленая зона Rf 0,75, зона кобальта окрашена в желтый цвет Rf 0,40, сине-фиолетовая зона характерна для никеля Rf 0,25.
Количественное определение меди, кобальта, никеля проводят непосредственно на хроматограмме с помощью спектрофотометра СФ-10 или СФ-14. Для этого на входе лучей в интегрирующий шар справа и слева по ходу лучей ставят плосковыпуклые линзы с оптической силой +10D, концентрируя световой пучок до размеров 6,5 на 15 мм. Проводят измерение величины отражения со всей площади окрашенной зоны. В качестве фона для сравнения берут две контрольные полоски бумаги и помещают в кюветы, расположенные справа и слева в нижней части интегрирующего шара. Записывают отражение фона. Затем вместо правого по ходу лучей эталона помещают измеряемый образец и записывают спектр отражения, по которому далее находят область минимального значения отражения каждого элемента. При этой длине волны вычисляют коэффициенты отражения (Rотр.) для меди, кобальта, никеля по формуле:
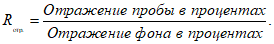
Для построения калибровочного графика готовят шкалу стандартов. При этом смесь стандартных растворов меди, кобальта и никеля наносят на фильтры, употребляемые для отбора пробы (табл. 31).
Таблица 31
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Смесь стандартных растворов никеля, кобальта, меди по 10 мкг, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Соляная кислота 7 н., мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание никеля, кобальта, меди, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Фильтры с нанесенными стандартными растворами помещают в тигли, добавляют 0,25 мл серной кислоты и содержимое выпаривают досуха. Пробы озоляют при 500 °C. Далее шкалу стандартов обрабатывают аналогично пробе.
Калибровочный график строят с применением метода Кортюма в координатах:
на оси ординат 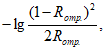
на оси абсцисс ![]()
Количественное определение меди, кобальта, никеля может быть также проведено методом стандартных серий путем сравнения интенсивности зон локализаций пробы со шкалой стандартов.
Пример расчета:
Анализируемая проба рубеаната никеля при длине волны 610 нм имеет величину отражения 65%, отражение фона составляет 100%.
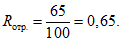
Вычисляется значение функции:
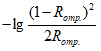
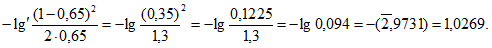
По калибровочному графику величина 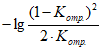 на оси ординат, равная 1,0269, соответствует на оси абсцисс величине -lgC = 0,18.
на оси ординат, равная 1,0269, соответствует на оси абсцисс величине -lgC = 0,18.
lgC = -0,18 = 1,82. По таблицам антилогарифмов находят значение концентрации С = 0,66 мкг.
ЦИНК
Металл серебристого цвета.
Плотность 7,14.
Температура плавления 419,505 °C, температура кипения 913 °C.
Не растворим в воде. Хорошо растворяется в слабых минеральных кислотах, концентрированных щелочах. Легко окисляется на воздухе, образуя окись цинка.
Соединения цинка токсичны. Растворимые соли цинка обладают прижигающим действием.
Окись цинка вызывает катаральное заболевание дыхательных путей и пищеварительных органов. Вдыхание паров окиси цинка связано с литейной лихорадкой.
ОПРЕДЕЛЕНИЕ ЦИНКА С СУЛЬФАРСАЗЕНОМ [2]
Принцип определения
При взаимодействии иона цинка с сульфарсазеном в среде винной, лимонной кислот и аммиака (pH 9,6) образуется комплексное соединение, окрашенное в оранжево-красный цвет.
Чувствительность — 0,5 мкг в определяемом объеме раствора.
Медь, свинец, никель, кобальт, марганец удаляют в процессе анализа, так как они мешают определению.
Железо3, ванадий4, молибден, алюминий, литий не мешают определению в количествах до 1 мг.
Реактивы
1. Исходный стандартный раствор с содержанием 1 мг цинка в 1 мл готовят растворением 0,025 г металлического (химически чистого) цинка в мерной колбе емкостью 25 мл; объем доводят до метки раствором соляной кислоты (1:1).
Стандартный раствор А с содержанием 0,1 мг цинка в 1 мл готовят следующим образом: 2,5 мл исходного стандартного раствора вносят в мерную колбу емкостью 25 мл, нейтрализуют 5% раствором едкого натра по лакмусу. Объем доводят до метки водой.
Рабочий стандартный раствор с содержанием 0,01 мг цинка в 1 мл готовят разбавлением водой раствора А в 10 раз.
2. Едкий натр, 5% раствор.
3. Соляная кислота, уд. вес 1,19 (1:1).
4. Диэтилдитиокарбамат натрия, 1% раствор.
5. Серная кислота, 0,1 н. раствор.
6. Аммиак, 5% раствор. 5 мл концентрированного аммиака помещают в мерную колбу емкостью 100 мл, доводят до метки водой. 10 мл раствора титруют 0,1 н. раствора серной кислоты в присутствии метилового оранжевого, до перехода желтой окраски в розовую. 1 мл 0,1 н. раствора серной кислоты соответствует 0,001703 г аммиака. Соответствующим разбавлением готовят 5% раствор аммиака.
7. Винная кислота, 10% раствор.
8. Лимонная кислота, 10% раствор.
9. Сульфарсазен, 0,02% водный раствор (свежеприготовленный).
10. Сульфат меди. Раствор с содержанием 1 мг меди в 1 мл готовят растворением 0,393 г сульфата меди в мерной колбе емкостью 100 мл, объем до метки доводят водой.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью 10 — 15 л/мин. протягивают через фильтр АФА или бумажный фильтр, укрепленный в патроне. Продолжительность отбора 20 — 30 мин.
Б. Для определения среднесуточной концентрации воздух со скоростью 2 — 5 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток.
Допустимо среднесуточный отбор проводить в один и тот же фильтр 6 — 12 раз с перерывом в 4 или 2 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в фарфоровую чашку, в которую вносят 10 мл раствора соляной кислоты 1:1, и кипятят в течение 5 мин.
Фильтр отжимают, промывают водой и удаляют. Промывные жидкости объединяют в фарфоровой чашке. В чашку вносят 200 мкг сульфата меди для соосаждения сопутствующих металлов (меди, свинца, кобальта, марганца, никеля), содержимое чашки упаривают досуха на водяной бане. Сухой остаток растворяют в 1,6 мл 5% раствора аммиака и добавляют 0,3 мл 1% раствора диэтилдитиокарбамата натрия. Перемешивают и добавляют 1,3 мл воды. Выделившийся осадок диэтилдитиокарбаматов металлов фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в которую вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см.
Для анализа берут 1,6 мл пробы, добавляют 1,1 мл воды, 0,8 мл 10% раствора винной кислоты, 0,2 мл 10% раствора лимонной кислоты, 0,3 мл 5% раствора аммиака и 1 мл 0,02% раствора сульфарсазена. Раствор перемешивают и интенсивность окраски измеряют при помощи фотометра в кювете с толщиной слоя 1 см при длине волны 500 нм по сравнению с контролем.
По предварительно построенному калибровочному графику устанавливают содержание цинка в анализируемом объеме пробы.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, которыми пользуются при отборе проб. Одновременно готовят контрольный фильтр (табл. 32).
Таблица 32
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
|
Содержание цинка, мкг |
0 |
0,5 |
1,0 |
3,0 |
5,0 |
7,0 |
10 |
Контрольный фильтр и фильтры, содержащие различное количество цинка, обрабатывают аналогично пробам. Шкала стандартов может быть использована для визуального определения. Ее готовят в колориметрических пробирках, как указано в табл. 32.
Расчет см. выше.
РТУТЬ
Серебристый жидкий металл.
Плотность 13,546. Температура плавления 38,87 °C, температура кипения 356,58 °C.
В воздухе может находиться в виде паров, причем концентрация ртути меняется с переменой давления и температуры.
Максимально возможная концентрация ртути: при 20 °C и давлении 0,0013 мм 15,2 мг/м3, при 30 °C и давлении 0,0029 мм 33,9 мг/куб. м, при 40 °C и давлении 0,0060 мм 70 мг/м3.
Пары ртути в 7 раз тяжелее воздуха.
Ртуть очень мало растворима в воде, растворяется в растворах хлористого натрия, азотной, серной кислотах, царской водке. Образует амальгамы с рядом металлов, оловом, цинком, свинцом и др.
Ряд соединений ртути под действием восстановителей легко разлагается с выделением металлической ртути.
Пары ртути и ее соединения ядовиты, вызывают нарушения функции нервной системы, почек, печени, обладают кумулятивным действием.
Предельно допустимая концентрация ртути металлической: среднесуточная 0,0003 мг/м3.
ОПРЕДЕЛЕНИЕ РТУТИ [6]
Принцип определения
При взаимодействии ртути с йодом образуется йодистая ртуть, которая с двуххлористой медью и сульфатом натрия образует взвесь, представляющую собой комплексное соединение 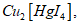 окрашенное в красный цвет. Одновременно образуется белая взвесь CuI, служащая фоном, благодаря чему осадок в присутствии ртути окрашен от желтовато-розового до красновато-оранжевого цвета.
окрашенное в красный цвет. Одновременно образуется белая взвесь CuI, служащая фоном, благодаря чему осадок в присутствии ртути окрашен от желтовато-розового до красновато-оранжевого цвета.
Чувствительность определения 0,02 мкг в определяемом объеме раствора. Определению мешает железо.
Реактивы
1. Йод кристаллический.
2. Йодид калия, х.ч.
3. Поглотительный раствор. Готовят из 3 г йодида калия, в который добавляют 3 — 5 мл воды и вносят 0,25 г йода. После растворения йода доводят объем дистиллированной водой до 1 л.
4. Исходный стандартный раствор с содержанием 100 мкг ртути в 1 мл готовят растворением 0,0135 г хлорида ртути или 0,0226 г йодида ртути в мерной колбе емкостью 100 мл. Объем до метки доводят поглотительным раствором.
Рабочий стандартный раствор с содержанием 0,1 мкг в 1 мл готовят разбавлением исходного в 1000 раз поглотительным раствором.
5. Хлорид меди, 7% раствор.
6. Сульфит натрия, х.ч., 3 н. раствор, 0,1 н. раствор, 37,8 г сульфита натрия растворяют в 100 мл воды. Титр 3 н. раствора сульфита устанавливают по 0,1 н. раствору йода. Раствор должен быть свежеприготовленный, так как сульфит натрия легко окисляется на воздухе в сульфат натрия. 0,1 н. раствор готовят разбавлением 3 н. раствора в 30 раз.
7. Смесь хлорида меди и сульфита натрия. Один объем 7% раствора хлорида меди вносят в мерный цилиндр, добавляют 5 объемов сульфита натрия. Осторожно перемешивают стеклянной палочкой до полного растворения осадка. Раствор должен быть прозрачным.
Отбор пробы
А. Для определения разовой концентрации воздух протягивают со скоростью 1 л/мин. через два последовательно соединенных поглотительных прибора Полежаева, наполненных по 1 мл поглотительного раствора.
Перед отбором проб к входному отверстию поглотительного прибора присоединяют компенсатор йода.
Компенсатор представляет собой сосуд, через который проходит впаянная стеклянная трубка с отверстием. Сбоку впаяна вторая трубка, через которую в сосуд перед отбором проб вносят 0,05 г кристаллического йода и отверстие закрывают. Отверстие в стеклянной трубке, проходящей внутри сосуда, служит для поступления йода в поглотительный раствор для уравнивания его концентрации.
Компенсатор присоединяют к системе поглотителей при помощи отрезка каучуковой трубки так, чтобы отверстие во внутренней трубке было обращено вверх (рис. 20).
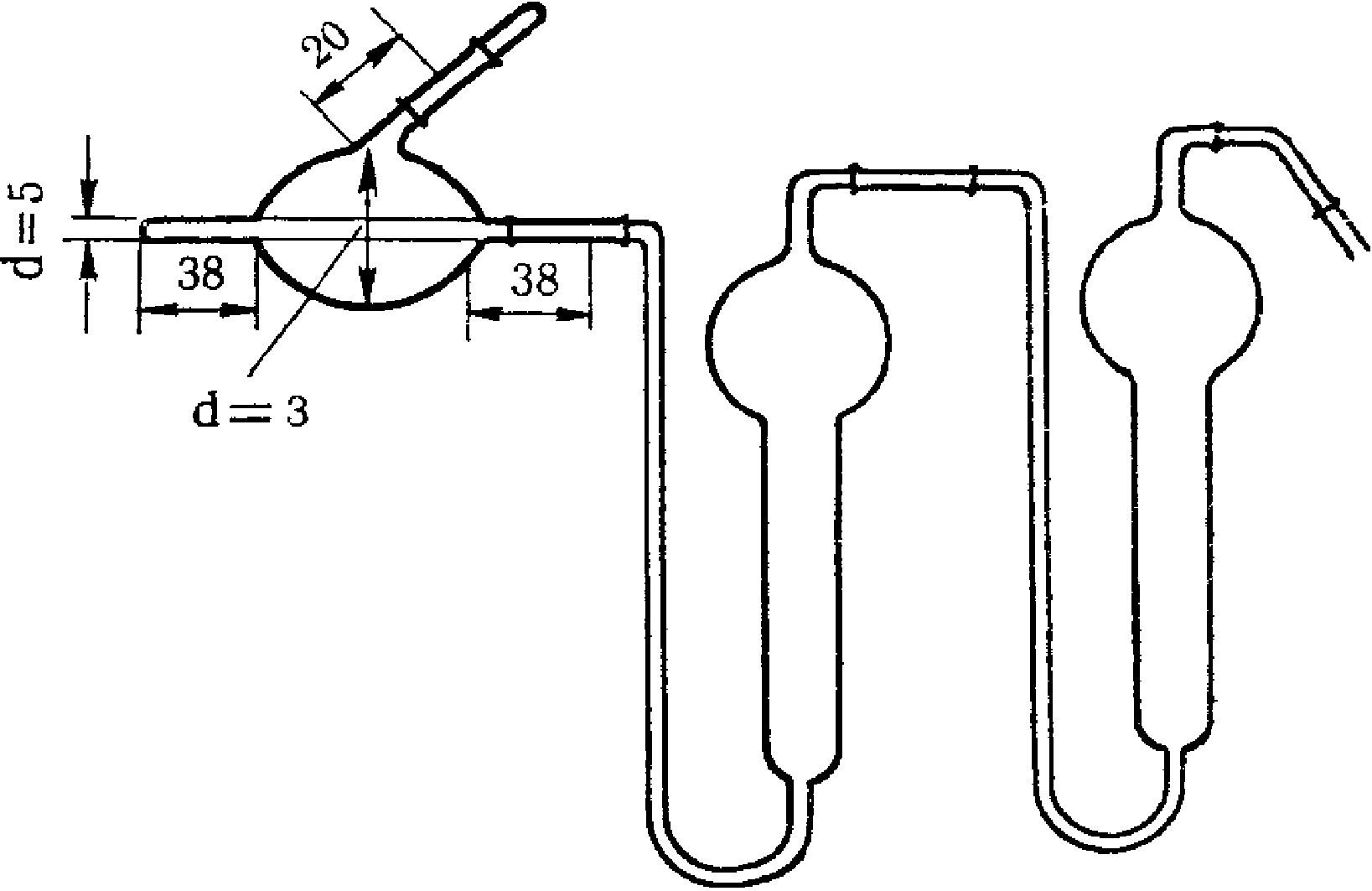
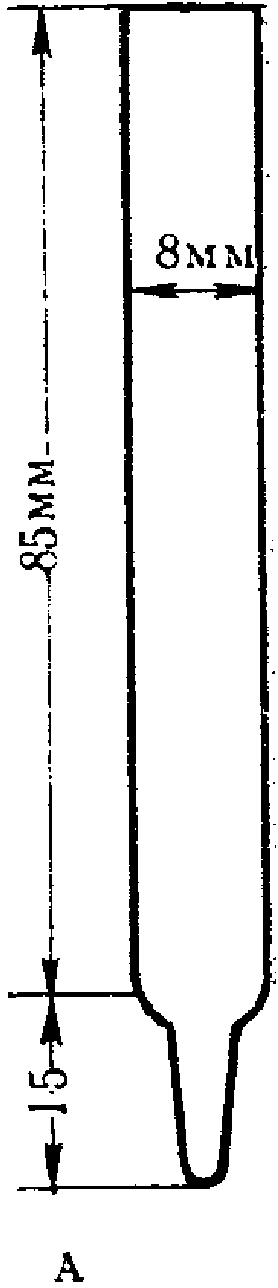
Рис. 20. Схема присоединения компенсатора йода
к поглотительным приборам. А — пробирка
для определения ртути
Продолжительность отбора пробы один час.
Б. Для определения среднесуточной концентрации воздух протягивают со скоростью 0,2 л/мин. через систему поглотителей, содержащих по 1 мл поглотительного раствора. Отмечают уровень жидкости.
Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 или 2 ч в одну и ту же систему поглотителей.
Ход анализа
Компенсатор йода отъединяют. Содержимое первого поглотительного прибора переносят в центрифужную пробирку через внутреннюю трубку компенсатора <1>. Промывают поглотительный прибор 1 мл воды и сливают ее в пробирку также через внутреннюю трубку компенсатора.
———————————
<1> Можно использовать пробирки, показанные на рис. 20.
Содержимое второго поглотительного прибора и 1 мл промывной воды переносят во вторую центрифужную пробирку.
В центрифужные пробирки, содержащие по 2 мл пробы, вносят одну каплю 0,1 н. раствора сульфита натрия для связывания йода. К бесцветному раствору пробы добавляют по 0,1 мл смеси хлорида меди и сульфита, содержимое пробирок перемешивают. Оставляют пробирки в покое на 10 — 15 мин. до появления взвеси. Далее центрифугируют в течение 5 мин. для осаждения полученной взвеси на дно пробирки и получения маленького очерченного круга.
Диаметр круга осадка в случае необходимости уменьшают вращательными движениями пробирки. Колориметрирование проводят по сравнению окраски кружков осадка с одновременно приготовленной шкалой (табл. 33). Сравнение удобно проводить при помощи зеркала, поставленного под углом 45° по отношению вертикально стоящих пробирок с пробами и стандартной шкалой.
Таблица 33
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1,0 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Вода, мл |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
|
Содержание ртути, мкг |
0 |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
Во все пробирки шкалы добавляют реактивы аналогично пробе.
Расчет см. выше.
СВИНЕЦ
Мягкий металл серебристого цвета.
Плотность 11,34. Температура плавления 327,3 °C. При нагревании расплавленного свинца до 400 — 500 °C в воздухе появляются пары, быстро окисляющиеся до окиси свинца.
Температура кипения 1751 °C.
Растворим в разбавленной азотной, концентрированной серной, уксусной кислотах. В воде незначительно растворим в присутствии CO2, NH3.
Известны окисные соединения свинца: PbO — глет или массикот, Pb3O4 — сурик, PbO2 — двуокись, Pb2O — закись и многочисленные его соединения.
Токсическое действие свинца и его соединений сходно.
Свинец обладает общеядовитым действием. Наблюдаются изменения в нервной системе, крови, сосудах. Обладает кумулятивным свойством. Различная токсичность соединений свинца объясняется степенью их растворимости.
Предельно допустимая концентрация свинца и его соединений (кроме тетраэтилсвинца) в пересчете на свинец: среднесуточная — 0,0007 мг/м3.
Предельно допустимая концентрация свинца сернистого: среднесуточная 0,0017 мг/м3.
ОПРЕДЕЛЕНИЕ СВИНЦА С ХРОМАТОМ КАЛИЯ [7]
Принцип определения
Метод может быть использован для определения окиси, двуокиси, сульфата, сульфита, сульфида, хлорида, ацетата, нитрата свинца, сурика, галенита и др.
При взаимодействии свинца с хроматом калия происходит образование хромата свинца, который определяют по степени помутнения пробы.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Соли бария мешают определению.
Реактивы
1. Стандартный раствор свинца. Исходный раствор с содержанием 1 мг/мл готовят растворением 1,5884 г перекристаллизованного нитрата свинца в мерной колбе емкостью 1 л в 3% растворе ацетата аммония или буферном растворе.
Разбавляя исходный раствор в 10 раз, получают стандартный раствор А с содержанием 100 мкг/мл. Рабочий стандартный раствор Б с содержанием 10 мкг/мл готовят разбавлением раствора А в 10 раз. Растворы А и Б готовят перед употреблением.
2. Уксусная кислота, 2 и 0,5% растворы.
3. Метиловый красный. 0,1 г метилового красного растворяют в 30 мл этанола и добавляют 50 мл воды.
4. Ацетат аммония, 3% раствор, подкисленный уксусной кислотой до pH 6,6 — 6,8. pH среды устанавливают по изменению окраски метилового красного или при помощи pH-метра.
5. Ацетат натрия, 1% раствор.
6. Этанол.
7. Буферная смесь 1% раствор ацетата натрия в 0,5% растворе уксусной кислоты.
8. Серная кислота, уд. вес 1,82 — 1,84, разбавленная 1:2.
9. Азотная кислота, уд. вес 1,4, разбавленная 1:2.
10. Смесь разбавленных кислот азотной и серной 1:5.
11. Хромат калия, 1% раствор.
Отбор пробы
А. Для определения разовой концентрации воздух со скоростью до 100 л/мин. протягивают через фильтр АФА, укрепленный в патроне.
Продолжительность отбора 30 мин. и более.
Б. Для определения среднесуточной концентрации воздух со скоростью 5 л/мин. протягивают через фильтр, укрепленный в патроне, непрерывно в течение суток.
Допустимо среднесуточный отбор проводить в один и тот же фильтр со скоростью 5 — 10 л/мин. с перерывами в 2 или 4 ч.
Ход анализа
Фильтр извлекают из патрона, обрезают края, переносят в фарфоровую чашку или тигель, смачивают 1 — 2 мл смеси кислот, нагревают на песчаной бане до образования твердого остатка. Пробу переносят в муфельную печь, прикрывая крышкой. Озоление проводят при температуре не выше 600 °C, поднимать температуру выше указанной не рекомендуется во избежание улетучивания сернокислого свинца. По озолении фарфоровую чашку (при закрытой крышке) вынимают из муфельной печи, охлаждают и вносят в нее 1 мл 3% раствора ацетата аммония или буферной смеси. Раствор фильтруют через воронку диаметром 2 — 3 см с оттянутым концом, в который вложен тампон, состоящий из волокон бумажного фильтра, уложенных длиной около 0,5 см. Чашку обмывают по 0,5 мл раствором ацетата аммония или буферной смесью. Растворы фильтруют и объем доводят до 2 мл, обмывая чашку и фильтруя сливы через ту же коронку. Можно сразу в чашку внести 2 мл смеси, провести растворение сульфата свинца и перенести раствор в центрифужную пробирку, обмыть чашку, общий объем довести до 3 мл. После центрифугирования для анализа брать 2 мл прозрачного раствора. В пробу вносят 0,1 мл 1% раствора хромата калия, перемешивают и через 20 — 30 мин. сравнивают интенсивность помутнения пробы со шкалой. Рассматривают на темном фоне. Одновременно с пробами готовят стандартную шкалу и контроль. Для этого на фильтры, употребляемые для отбора проб, наносят различные количества стандартного раствора Б (табл. 34).
Таблица 34
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл (Б) |
0 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
|
Содержание свинца, мкг |
0 |
2 |
3 |
4 |
5 |
6 |
Контрольный фильтр и фильтры, содержащие различные количества свинца, помещают в чашки и обрабатывают аналогично пробам.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СВИНЦА С ДИТИЗОНОМ [5]
Принцип определения
При взаимодействии неорганических соединений свинца с дитизоном образуется дитизонат, окрашенный в красный цвет, растворимый в хлороформе и четыреххлористом углероде.
Чувствительность определения 2 мкг в анализируемом объеме. Железо в количествах до 100 мкг, медь 50 мкг, цинк до 500 мкг не мешают определению.
Реактивы <1>
———————————
<1> Все реактивы должны быть приготовлены на бидистиллированной воде.
1. Стандартный раствор свинца. Исходный раствор с содержанием 1 мг/мл готовят путем растворения 1,5984 г перекристаллизованного нитрата свинца в мерной колбе емкостью 1 л в 1% растворе азотной кислоты. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят разбавлением исходного в 100 раз.
2. Азотная кислота, уд. вес 1,4, х.ч., свободная от свинца, разбавленная 1:1; 2 н. раствор, последний готовят, разбавляя с 145 мл азотной кислоты, уд. вес 1,4 до 1 л.
Азотная кислота, 1% раствор; 15 мл концентрированной азотной кислоты разбавляют водой до 1 л.
3. Соляная кислота, уд. вес. 1,19, х.ч., не содержащая свинца, бидистиллированная, 1 н. раствор.
4. Винная кислота, х.ч., 10% раствор.
5. Виннокислый калий-натрий, х.ч.
6. Аммиак, уд. вес 0,91, х. ч., свободный от свинца, 1% раствор, 0,02 н. раствор.
7. Цианид калия, х.ч.
8. Сульфит натрия.
9. Буферный раствор: в 350 мл аммиака растворяют 10 г цианида калия, 15 г сульфита натрия и 15 г виннокислого калия-натрия.
10. Хлорид кальция безводный.
11. Хлороформ или четыреххлористый углерод. Очищают промыванием бидистиллированной водой, затем разбавленной соляной кислотой, 0,02 н. раствором аммиака. Осушают безводным хлоридом кальция и через 15 ч отгоняют. Аналогично регенерируют употребленный растворитель.
12. Дитизон. 30 мл дитизона растворяют в 1 л хлороформа или четыреххлористого углерода. Раствор дитизона должен быть приготовлен за 2 дня перед применением, его хранят в темном месте при охлаждении. Перед применением проверяют чистоту раствора дитизона. В пробирку вносят 5 мл раствора дитизона, промывают 5 мл 1% раствора аммиака, сильно встряхивая в течение 1 мин. Раствор применим, если слой органического растворителя бесцветный или окрашен в слабо-желтый цвет. Если раствор дитизона загрязнен продуктами окисления, то слой органического растворителя окрашен в желтый цвет и его подвергают очистке. Для этого 40 мг дитизона растворяют в 250 мл хлороформа или четыреххлористого углерода, переносят в делительную воронку емкостью 1 л, встряхивают в течение 15 мин. Далее добавляют 500 мл 0,02 н. раствора аммиака и вновь встряхивают в течение 5 — 10 мин. Оставляют в покое до расслоения жидкостей. Дитизон переходит в водный слой, а продукты окисления остаются в слое органического растворителя. Слой органического растворителя отделяют, а аммиачный слой несколько раз промывают растворителем, добавляя его по 10 — 12 мл. После очистки аммиачный слой должен иметь чисто зеленую окраску. Затем к аммиачному слою добавляют 125 мл органического растворителя и 12 мл 1 н. раствора соляной кислоты, встряхивают до тех пор, пока дитизон не перейдет в слой органического растворителя. Отделяют органический слой, содержащий дитизон, и промывают его бидистиллированной водой; затем доводят растворителем до 1 л и добавляют 5 мл воды, насыщенной сернистым ангидридом. Раствор хранят в стеклянной таре из темного стекла при +4 °C. Раствор устойчив в течение нескольких месяцев.
Применяемая посуда (делительные воронки, мерные колбы и др.) должна быть приготовлена из твердого химического стекла (типа Ена). Перед употреблением ее следует тщательно промыть горячим концентрированным раствором щелочи, горячей азотной кислотой, бидистиллированной водой до нейтральной реакции.
Фарфоровые чашки также необходимо предварительно промыть, как и стеклянную посуду, и проверить их на отсутствие свинца.
Отбор пробы см. выше.
Ход анализа
Фильтр извлекают из патрона, обрезают края, помещают в маленькую фарфоровую чашку, добавляют 10 мл азотной кислоты 1:1 и нагревают на кипящей водяной бане в течение 10 мин. При наличии в пробе PbO2 и Pb3O4 добавляют еще 10 мл концентрированной соляной кислоты и снова нагревают в течение 10 мин. Раствор отфильтровывают. Фильтр промывают 2 раза по 5 мл горячей 2 н. азотной кислотой, а затем горячей водой. Фильтрат упаривают в фарфоровой чашке на водяной бане. Сухой остаток растворяют в 5 мл горячей 10% винной кислоты и 2 мл 2 н. азотной кислоты. Раствор переносят в мерную колбу емкостью 25 мл, чашку несколько раз обмывают горячей водой. После охлаждения объем доводят до метки бидистиллированной водой.
В чистую сухую делительную воронку вносят 12,5 мл пробы, добавляют 37,5 мл буферного раствора и 12,5 мл раствора дитизона. Содержимое воронки встряхивают в течение 3 мин. После расслоения жидкостей конец делительной воронки высушивают, промывают путем спускания небольшого количества пробы. Пробу переносят в кювету и фотометрируют при длине волны 520 нм (зеленый светофильтр), кювета имеет толщину слоя 2 или 1 см по сравнению с контролем (бледно-зеленым раствором).
Содержание свинца в анализируемом объеме устанавливают по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры, употребляемые для отбора. Одновременно готовят и контроль, как указано в табл. 35.
Таблица 35
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,3 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание свинца, мкг |
0 |
2 |
3 |
4 |
6 |
8 |
10 |
Контрольный фильтр и фильтры, содержащие различные количества свинца, помещают в фарфоровые чашки и обрабатывают аналогично пробам.
Расчет см. выше.
ЛИТЕРАТУРА
1. Кузьмичева М.Н. К вопросу определения ванадия в воздухе. Гиг. и сан., 1966, 2.
2. Крылова Н.А. Определение цинка в воздухе с сульфарсазеном. Гиг. и сан., 1970, 3.
3. Логинов М.Е. Определение хрома. В кн.: М.В. Алексеева. Определение атмосферных загрязнений. М., «Медгиз», 1963.
4. Маркина Н.А. Хроматографическое разделение микроколичеств меди, кобальта, никеля в присутствии железа в атмосферном воздухе. Гиг. и сан., 1971, 9, 11.
5. Определение неорганических соединений свинца по реакции с дитизоном. Определение соединений двух- и трехвалентного хрома с дифенилкарбазидом. В сб. СЭВ: Унифицированные методы определения атмосферных загрязнений. М., 1970.
6. Полежаев Н.Г. К методике определения ртути в атмосферном воздухе. Гиг. и сан., 1956, 6.
7. Хрусталева В.А. Определение органического и неорганического свинца в воздухе гаражей. Гиг. и сан., 1952, 6.
8. Хрусталева В.А. Определение аэрозолей двуокиси марганца в воздухе производственных помещений. Гиг. и сан., 1951, 10.
Глава VII
ОКИСЬ УГЛЕРОДА, БЕНЗИН, УГЛЕВОДОРОДЫ, СИНИЛЬНАЯ КИСЛОТА
ОКИСЬ УГЛЕРОДА
Бесцветный газ, без запаха и вкуса. Плотность по отношению к воздуху 0,967. Температура кипения — 190 °C. Коэффициент растворимости в воде 0,2489 (20°), 0,02218 (30°), 0,02081 (38°), 0,02035 (40°). Вес 1 л газа при 0 °C и 760 мм рт. ст. 1,25 г. Входит в состав различных газовых смесей, коксового, сланцевого, водяного, древесного, доменного газов, выхлопных газов автотранспорта и др.
Горит синеватым пламенем. Смесь, состоящая из 1 объема кислорода и 2 объемов окиси углерода, взрывается при внесении пламени. Окись углерода является продуктом неполного сгорания веществ, в которых содержится углерод.
Обладает восстанавливающими свойствами, способна к реакциям присоединения; активность ее повышается в присутствии катализаторов.
Окись углерода токсична, вытесняет кислород из гемоглобина крови, образуя при этом карбоксигемоглобин; оказывает непосредственное действие на клетки, при этом уменьшается потребление кислорода тканями; влияет на углеводный обмен, обмен фосфора, действует на центральную нервную систему. Действие окиси углерода выражается в потере сознания, судорогах, одышке, удушье.
Предельно допустимые концентрации: максимальная разовая — 3 мг/м3, среднесуточная — 1 мг/м3.
ОПРЕДЕЛЕНИЕ ОКИСИ УГЛЕРОДА С ЙОДНОВАТЫМ АНГИДРИДОМ [2]
Принцип определения
При взаимодействии окиси углерода с йодноватым ангидридом при 130 — 150 °C происходит окисление ее до углекислоты. Последняя улавливается раствором гидрата окиси бария, избыток которого титруется соляной кислотой.
Чувствительность метода: при титровании 0,01 н. раствором соляной кислоты — 0,0014 мг (1,4 мкг), при титровании 0,005 н. раствором соляной кислоты — 0,0007 мг (0,7 мкг).
Аппаратура
1. Прибор для определения окиси углерода (рис. 21). Прибор смонтирован на деревянной основе и состоит из ряда очистительных колонок и колонки с йодноватым ангидридом, соединенных последовательно. На обратной стороне прибора расположены две очистительные колонки (9), одна наполнена пемзой, которая пропитана концентрированной серной кислотой для поглощения влаги, вторая — кусочками едкого натра, поглощающего углекислоту. На лицевой стороне прибора укреплены манометр (4) и V-образные трубки (5) высотой около 20 см и диаметром 1 см. V-образные трубки соединены между собой при помощи небольших дугообразных трубок (3).
Для очистки исследуемого воздуха от углекислоты первые две V-образные трубки заполнены кусочками едкого натра в смеси с натронной известью в соотношении 3:1. Последующие заполнены силикагелем, поглощающим предельные и непредельные углеводороды, а последняя трубка также содержит кусочки едкого натра. Верхние части всех трубок на расстоянии 2 — 5 см заполнены стеклянной ватой.
Окисление окиси углерода до углекислоты происходит в V-образной трубке (6), наполненной на 3/4 объема йодноватым ангидридом и помещенной в электрическую печь (1). Электрическая печь изготовлена из шамотной глины в смеси с волокнистым асбестом, внутри ее проложена обмотка из нихромовой проволоки, обеспечивающая обогрев печи до 200°. Контроль за температурой осуществляется при помощи реостата (2). В печи при температуре 140 — 150 °C, измеряемой термометром (8), при действии йодноватого ангидрида происходит окисление углерода до углекислоты. Выделяющийся при этом йод улавливают кристаллическим йодидом калия, который помещен в поглотительном сосуде (7). Этот сосуд, снабженный каучуком (10) и зажимом (11), защищают от действия света. Йодид калия по мере пожелтения заменяют свежим. Для поглощения йода можно применять и опилки электролитической меди. Прибор должен быть герметичен.
2. Поглотительные сосуды (см. рис. 21, А).
3. Подставки к поглотительным сосудам (см. рис. 21, Б).
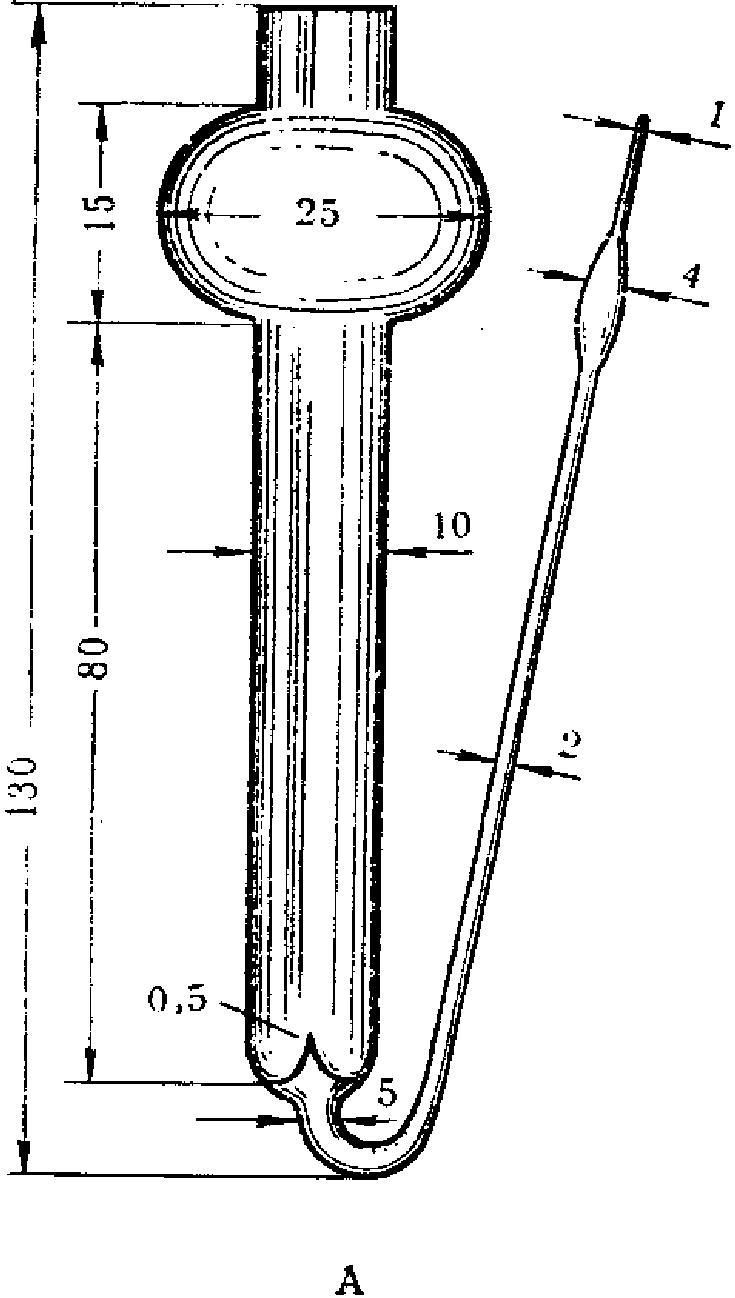
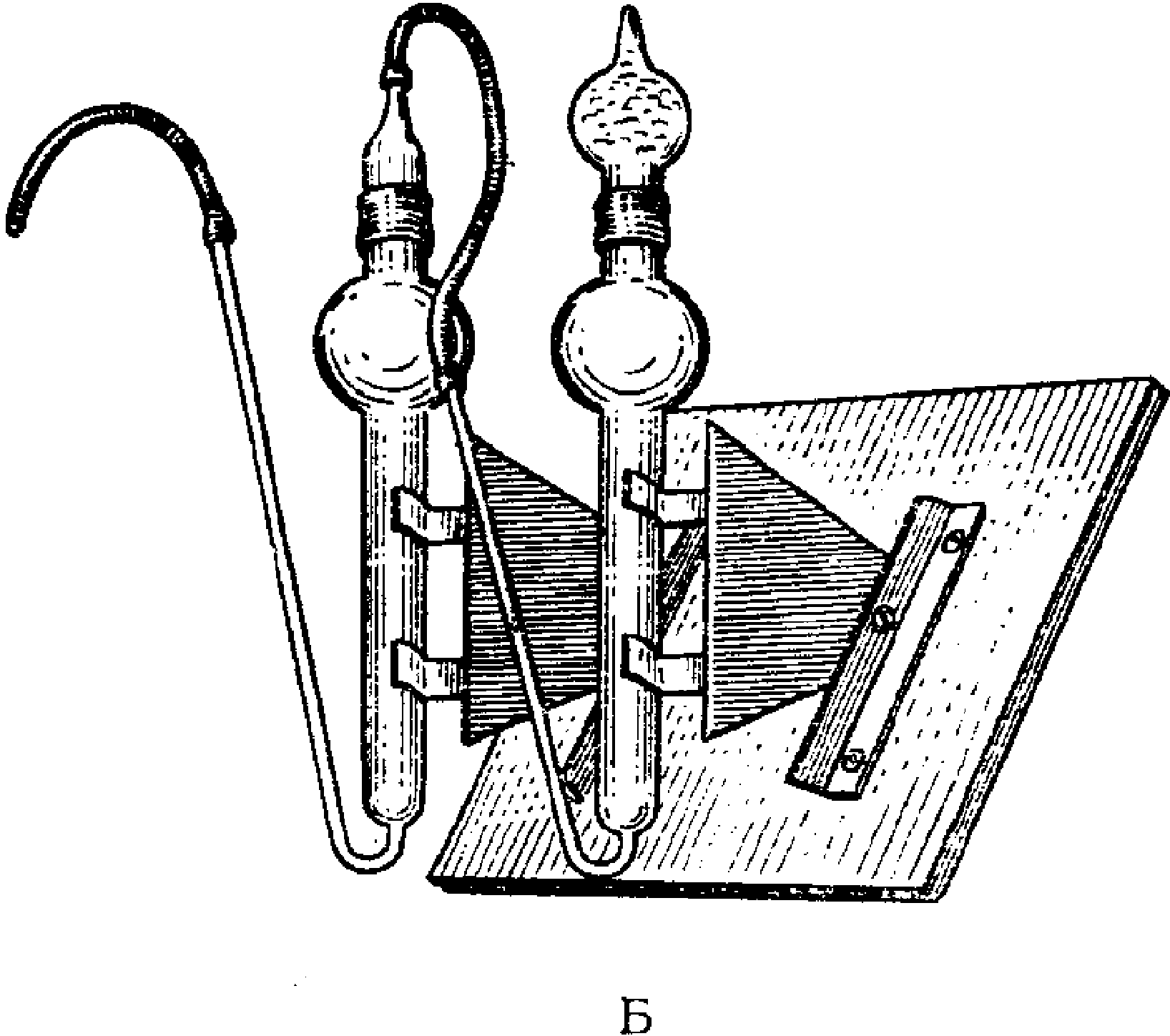
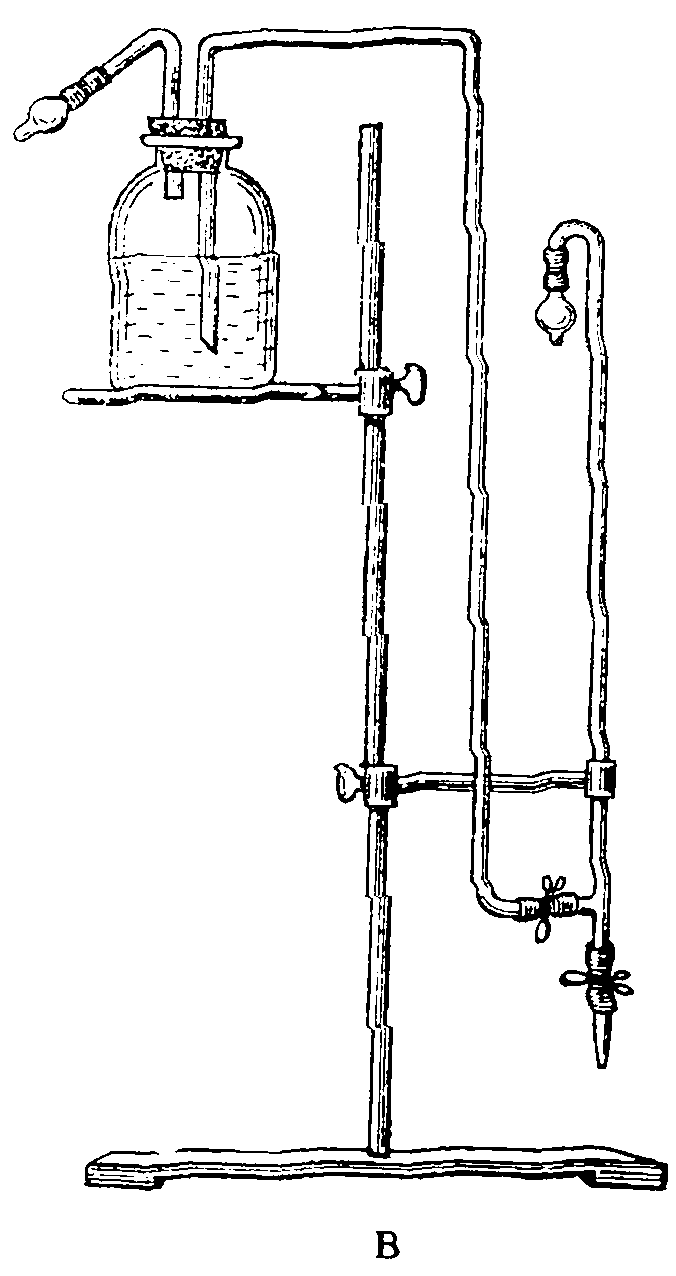
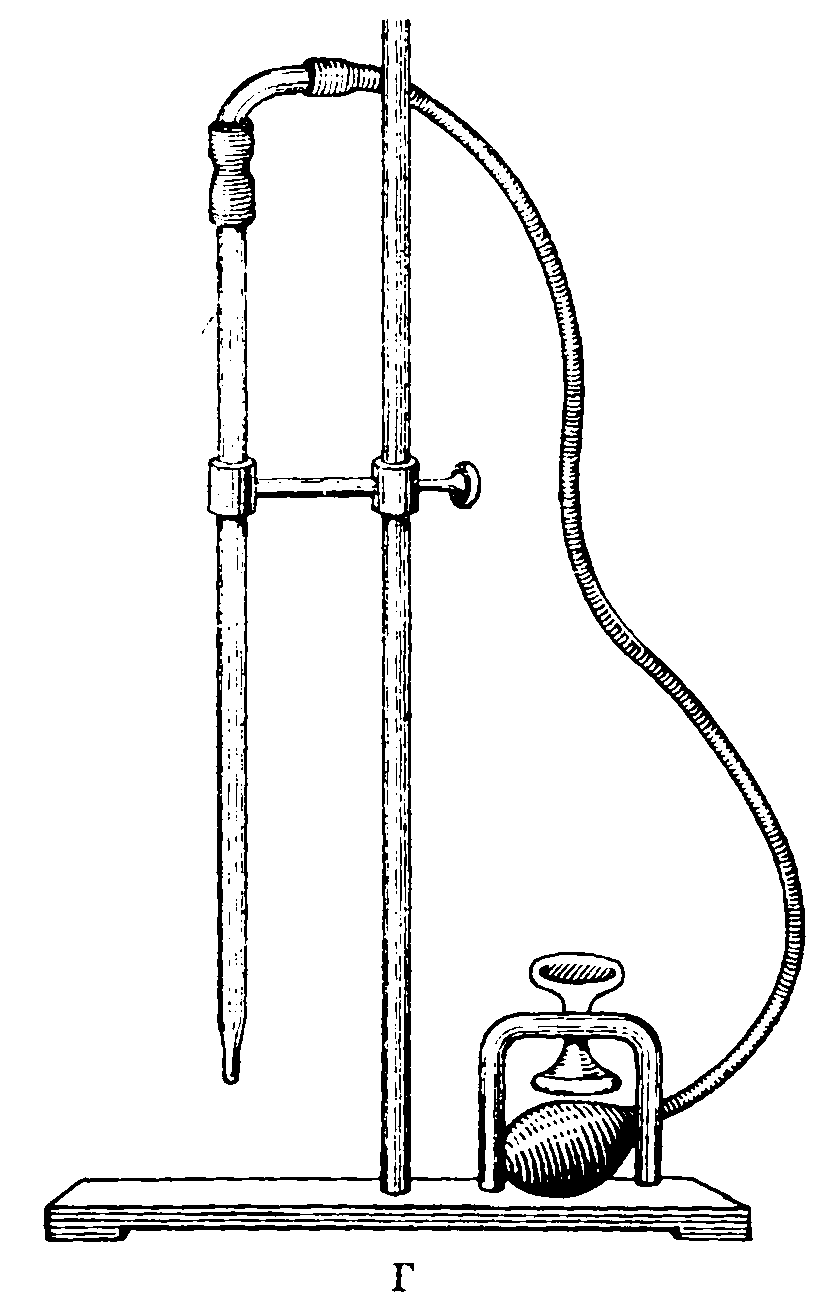
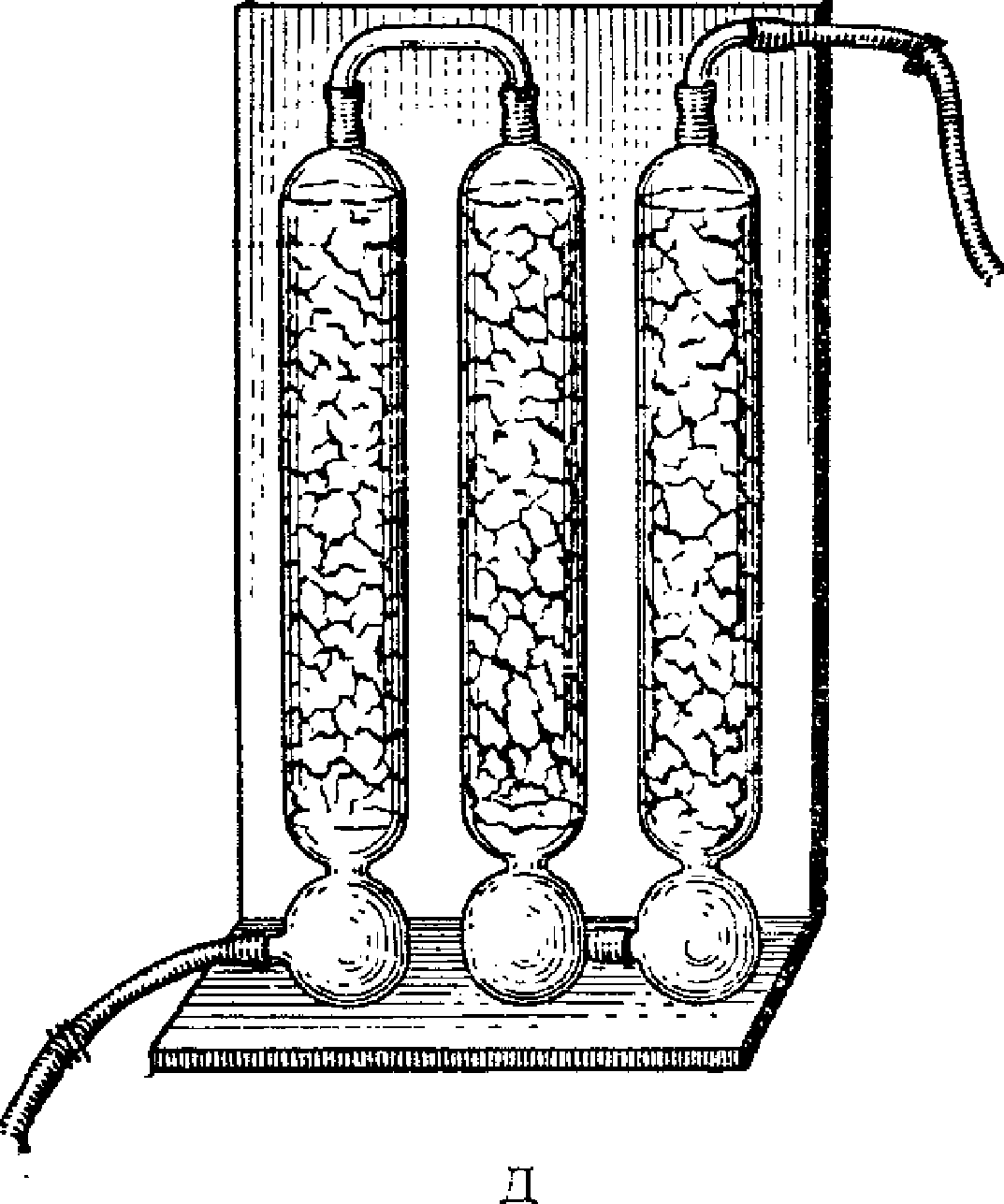
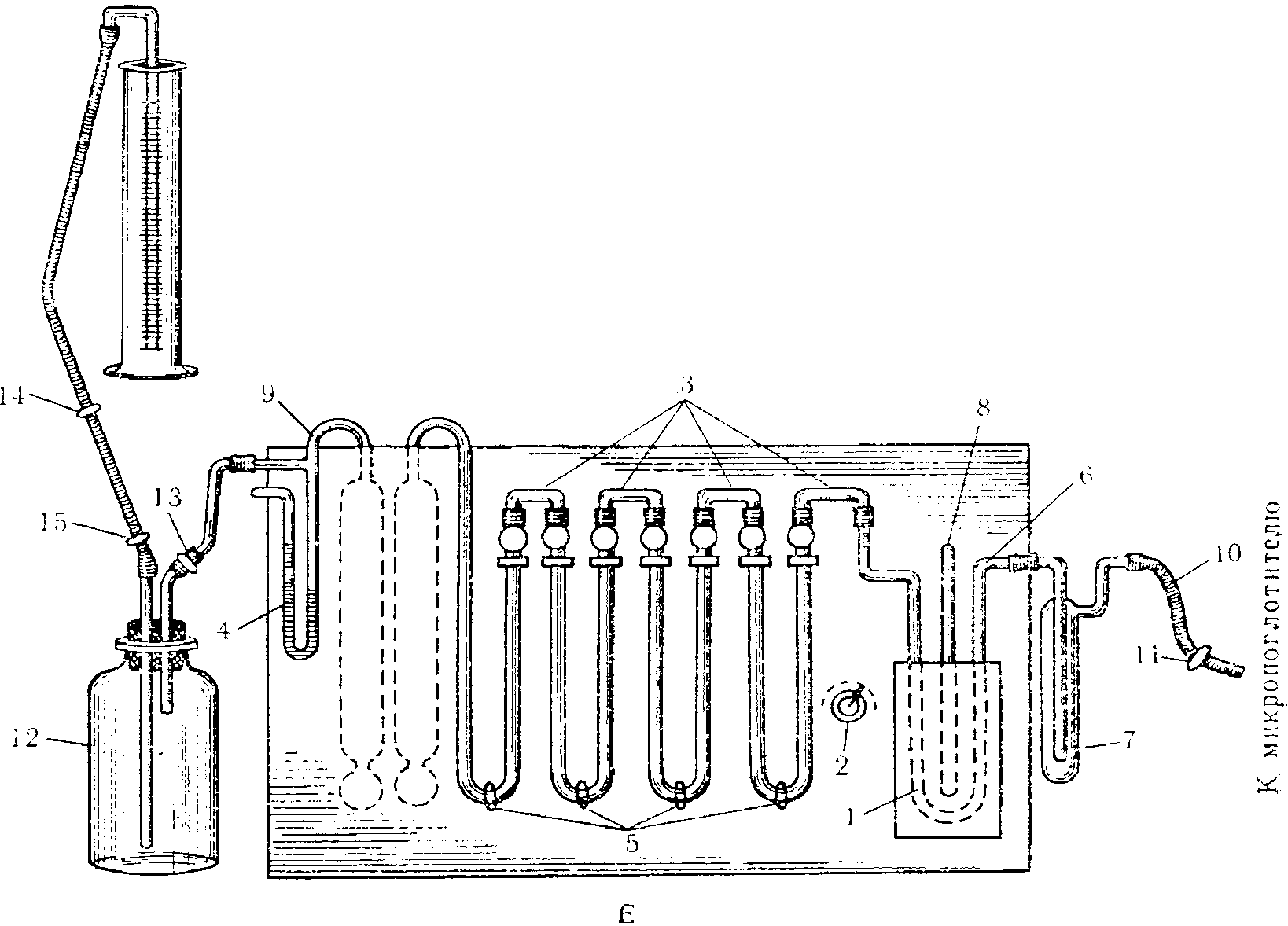
Рис. 21. Прибор для определения окиси углерода с йодноватым
ангидридом и отдельные детали к нему А — поглотительный
сосуд; Б — подставка к поглотительным сосудам; В — установка
для наполнения поглотительных сосудов; Г — пневматическая
бюретка; Д — очистительная система; Е — общий вид
4. Аспиратор, составленный из бутылей емкостью 5 — 6 л.
5. Установка для наполнения поглотительных сосудов гидратом окиси бария. Состоит из бутыли емкостью 2 — 3 л для раствора гидрата окиси бария, наполнительной бюретки емкостью 2 мл с делениями 0,01 мл. Верхний конец бюретки защищен трубкой с натронной известью (см. рис. 21, В). К установке приложены две очистительные колонки (см. рис. 21, Д), наполненные натронной известью в смеси с кусочками едкого натра, и пневматическая микробюретка (см. рис. 21, Г). Последняя представляет собой пипетку емкостью 2 мл с делением 0,01 мл. Нижний ее конец оттянут в капилляр диаметром не более 0,1 см. Верхняя часть микробюретки соединена с каучуковой трубкой длиной около 50 см, которая в свою очередь соединена с резиновым баллоном емкостью 20 — 30 мл, помещенным между пластинкой микрометрического винта и деревянной подставкой. При погружении кончика микробюретки в жидкость и расширении баллона проводят засос жидкости в бюретку. Обратное движение винта способствует вытеснению жидкости из микробюретки.
6. Бутыли для отбора проб воздуха емкостью 2 — 3 л, в которые вставлены пробки с двумя стеклянными трубками. Одна из них доходит почти до дна, другая — короткая. На верхние концы трубок надеты каучуковые трубки, закрытые стеклянными заглушками и винтовыми зажимами (см. рис. 21) (12, 13, 14, 15).
В настоящее время завод «Химлаборпосуда» изготавливает прибор ПОУ для определения окиси углерода по МРТУ 42-2068-20.
Реактивы
1. Соляная кислота 0,01 н. раствор, 0,005 н. раствор (точно приготовленный из фиксанала).
2. Гидрат окиси бария, 0,01 и 0,005 н. раствор. Готовят растворением 4,5 г гидрата окиси бария (кристаллы должны быть прозрачны, без налета углекислого бария) и 5 г хлорида бария в колбе емкостью 2 л. Колбу закрывают пробкой, в которую вмонтированы две стеклянные трубки, одна доходит почти до дна, другая короткая. Концы трубок защищают от влияния углекислоты натронной известью. Содержимое колбы взбалтывают и оставляют в покое на 1 — 2 суток, до получения прозрачного раствора. Прозрачный раствор из колбы сифонируют в бутыль емкостью 2 — 3 л, которую предварительно и в момент сифонирования продувают током воздуха, лишенным углекислоты. Бутыль с раствором гидрата окиси бария закрывают пробкой, в которую вмонтированы две трубки — длинная и короткая. Короткую трубку защищают натронной известью в смеси с кусочками едкого натра.
От влияния углекислоты длинную трубку соединяют с бюреткой. Верхняя часть бюретки также защищена натронной известью.
3. Хлорид бария.
4. Фенолфталеин, 0,5% раствор в 50% растворе этанола.
5. Этанол.
6. Натронная известь гранулированная.
7. Едкий натр, 40% раствор.
8. Пемза гранулированная.
9. Хлорид натрия, 26% раствор.
10. Силикагель, размер зерен 2 — 4 мм, активированный при 200 °C в течение 30 — 40 мин.
11. Серная кислота, уд. вес 1,84.
12. Йодид калия.
13. Йодноватый ангидрид, гранулированный. Порошкообразный йодноватый ангидрид или йодноватую кислоту помещают в фарфоровую чашку, увлажняют водой. Чашку ставят на водяную баню и по мере испарения воды, помешивают стеклянной палочкой. Образующиеся гранулы доводят до размера 2 — 3 см, подсушивают в сушильном шкафу при 105 °C в течение 1 — 2 ч. V-образную трубку наполняют 8 — 10 г гранулированного йодноватого ангидрида, помещают ее в электрическую печь прибора, соединяют с очистительной системой и продувают воздухом в течение нескольких часов. Печь нагревают, постепенно повышая температуру. Когда выделение влаги и йода прекратится, температуру повышают до 200° и продувают еще в течение 1 1/2 — 2 ч. Йодноватый ангидрид сохраняет восстановительную способность в течение длительного времени; 1 г йодноватого ангидрида способен окислить 400 г окиси углерода. Окислительную способность йодноватого ангидрида проверяют на пробах воздуха с известным содержанием окиси углерода.
14. Медь электролитическая. Электролитическую медь просеивают через сито или освобождают от пыли путем отмучивания в этиловом эфире. Эфир отделяют, опилки высушивают при комнатной температуре. Затем медь помещают в V-образную трубку, прикрывают небольшим слоем стеклянной ваты и продувают воздухом в течение 30 мин., нагретым не выше 100 °C и очищенным от углекислоты.
Отбор пробы
А. Для определения разовой концентрации воздух отбирают в бутыль емкостью 2 — 3 л, закрытую резиновой пробкой, в которую вмонтированы две стеклянные трубки — длинная и короткая. Объем бутыли заполняют 26% раствором хлорида натрия.
При отборе к длинному концу трубки присоединяют каучуковую трубку длиной 40 — 50 см и выливают раствор хлорида натрия со скоростью около 100 мл/мин. При этом исследуемый воздух поступает в бутыль через короткую трубку. Продолжительность отбора 20 — 30 мин. По окончании отбора отверстия трубок закрывают стеклянными заглушками и винтовыми зажимами. Отбор проб может быть проведен в пипетки емкостью 0,5 — 1 л.
Б. Для определения среднесуточной концентрации пробу отбирают в течение 24 ч в аспиратор емкостью 3 л, наполненный 26% раствором хлорида натрия. Скорость отбора 0,1 л/ч, через 24 ч в верхней бутыли аспиратора должно быть около 0,5 л раствора хлорида натрия.
Ход анализа
а) Подготовка прибора. Перед началом анализа устанавливают соотношение растворов гидрата окиси бария и соляной кислоты. Для этого поглотительный сосуд продувают в течение 2 мин. током очищенного воздуха, затем ставят его вертикально под кончик бюретки, которая наполнена гидратом окиси бария. В токе очищенного воздуха спускают из бюретки в сосуд 2 мл раствора и добавляют каплю фенолфталеина. Скорость поступления очищенного воздуха регулируют так, чтобы жидкость не попадала в широкую часть поглотительного сосуда. Наполнив один поглотитель, при помощи каучуковой трубки соединяют широкий конец его с капиллярной трубкой второго поглотительного сосуда. Так же в токе очищенного воздуха вносят во второй поглотитель 2 мл раствора гидрата окиси бария. Таким образом, получают два последовательно соединенных поглотительных прибора, наполненных по 2 мл раствора гидрата окиси бария, через который проходит ток очищенного воздуха. Не прекращая ток воздуха, начинают титрование со второго поглотительного сосуда. Титрование проводят раствором соляной кислоты до обесцвечивания раствора. Далее второй поглотитель отъединяют и титруют первый. На 2 мл 0,01 н. раствора гидрата окиси бария должно пойти примерно 2 мл (не более) 0,01 н. раствора соляной кислоты. Результаты титрования каждого поглотительного сосуда не должны отличаться между собой более чем на 0,01 мл соляной кислоты.
б) Контрольный анализ. Для получения воздуха, не содержащего окиси углерода, перед прибором включают систему, состоящую из хлоркальциевой трубки, наполненной хлоридом кальция, последовательно соединяют с V-образной трубкой, наполняют йодноватым ангидридом и помещают в дополнительную печь.
Дополнительную печь и печь прибора нагревают до 130 — 150 °C. Наполняют два поглотительных прибора раствором гидрата окиси бария аналогично описанному выше. Поглотительные приборы присоединяют к прибору. Через всю систему протягивают 1 л воздуха в течение часа. Поглотительные приборы с раствором гидрата окиси бария устанавливают почти в горизонтальном положении с тем, чтобы воздух проходил через раствор непрерывной цепочкой. При прохождении воздуха через первую трубку с йодноватым ангидридом имеющаяся окись углерода полностью окисляется до углекислого газа, которая далее улавливается в очистительной системе прибора. После прохождения 1 л воздуха через поглотительные приборы, объем которого регистрируется специальным цилиндром, закрывают зажим непосредственно перед прибором и наблюдают прекращение потока воздуха через поглотительные приборы.
Затем при помощи микробюретки титруют раствор гидрата окиси бария 0,01 н. раствором соляной кислоты. Титр раствора гидрата окиси бария получается более низкий, чем при непосредственном титровании раствора. Снижение обычно не превышает 0,02 мл 0,01 н. раствора соляной кислоты. Контрольное определение повторяют до получения совпадающих результатов.
в) Анализ проб проводят аналогично контрольному. Но при этом отключают дополнительную очистительную систему от прибора, служащую для получения воздуха, не содержащего окиси углерода. К прибору присоединяют емкость с пробой, пропускают через прибор 500 мл исследуемого воздуха (для заполнения системы). Наполняют и присоединяют к прибору поглотительные приборы и протягивают 1 л исследуемого воздуха в течение 50 — 60 мин. Контрольный анализ проводят перед каждым определением.
Расчет ведут по формуле:
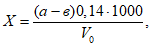
где: X — концентрация окиси углерода в миллиграммах на 1 м3 воздуха; а — количество 0,01 н. раствора соляной кислоты, израсходованное на титрование гидрата окиси бария при контрольном анализе; в — количество 0,01 н. раствора соляной кислоты, израсходованное на титрование гидрата окиси бария при анализе пробы; V0 — количество исследуемого воздуха (в литрах), приведенное к нормальным условиям; 1000 — множитель для выражения концентрации в миллиграммах на 1 м3 воздуха; 0,14 — количество окиси углерода, соответствующее 1 мл точно 0,01 н. раствору соляной кислоты (в миллиграммах); 0,007 — количество окиси углерода, соответствующее 1 мл точно 0,005 н. раствора соляной кислоты.
ОПРЕДЕЛЕНИЕ ОКИСИ УГЛЕРОДА НА ГАЗОАНАЛИЗАТОРЕ [3]
Принцип определения
При сжигании окиси углерода на платиновой спирали, нагретой до температуры красного каления (600°), образуется углекислота, которую поглощают раствором гидрата окиси бария. Количество углекислоты определяют по разности титрования избытка гидрата окиси бария раствором соляной кислоты до сжигания пробы и после него.
Чувствительность определения при титровании 0,01 н. раствором соляной кислоты 0,0014 мг (1,4 мкг).
При титровании 0,005 н. раствором соляной кислоты 0,0007 мг (0,7 мкг).
Определению мешает метан.
Газоанализатор состоит из очистительной и аналитической систем. Схема анализатора приведена на рис. 22.
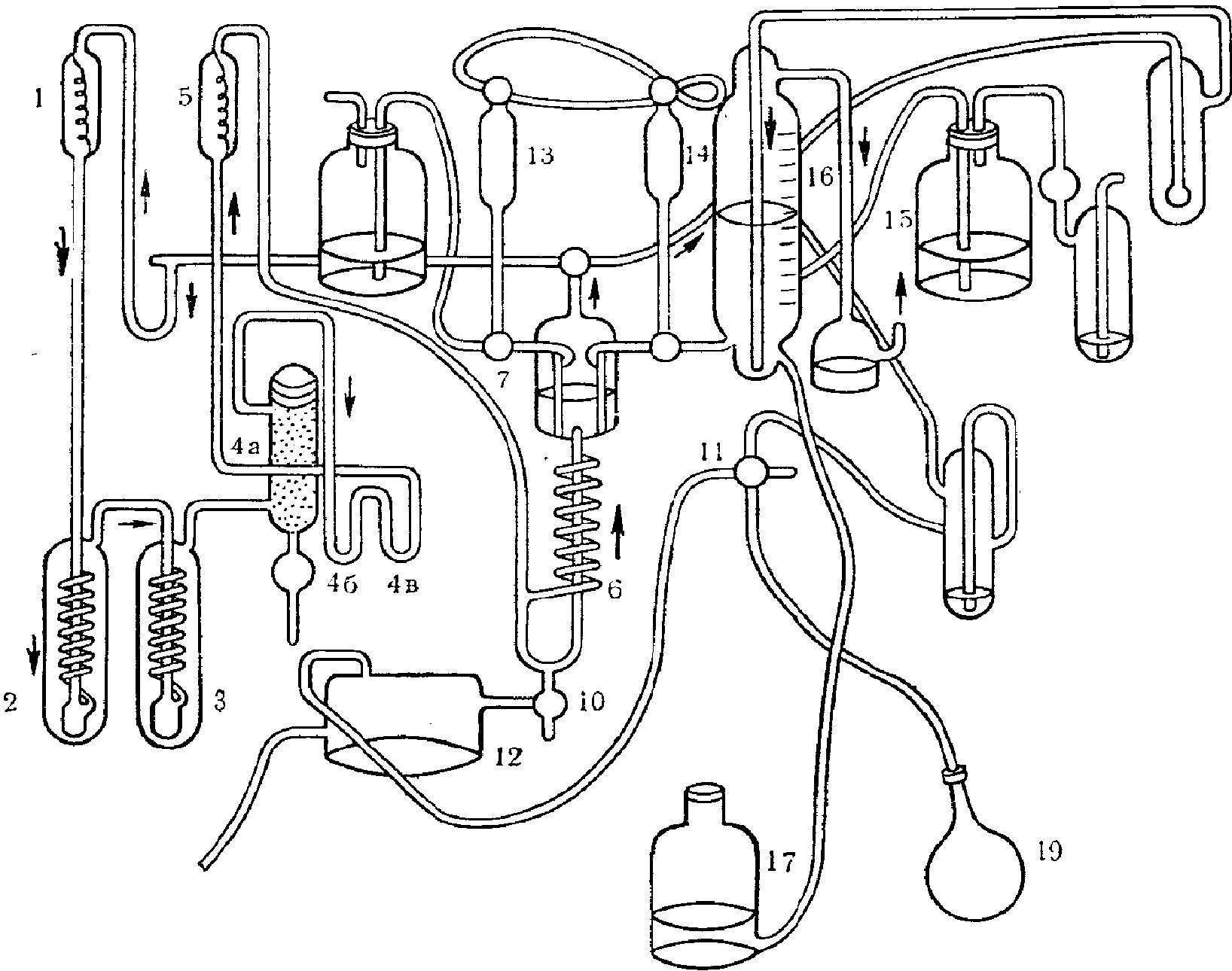
Рис. 22. Схема титрометрического газоанализатора
Очистительная система предназначается для поглощения углеводородов и двуокиси углерода из воздуха. Она состоит из колонки предварительного сожжения органических веществ воздуха (1), двух змеевиков-барботеров (2, 3), заполненных 40% раствором щелочи, патрона с хлоридом кальция (4а), двух V-образных трубок (4б, 4в); трубка 4б наполнена пемзой, смоченной серной кислотой удельного веса 1,82 — 1,84; трубка 4в — силикагелем.
В колонку предварительного сожжения впаяна спираль из платиновой проволоки длиной 70 мм и сечением 0,3 мм. Колонка обеспечивает полное сожжение горючих примесей в воздухе до двуокиси углерода, поглощаемой в барботерах с 40% раствором щелочи. Каждый из барботеров представляет собой сосуд, в который впаян змеевик.
В аналитическую часть входят следующие детали:
1. Колонка для сожжения окиси углерода по конструкции не отличается от колонки очистительной системы. Назначение колонки — каталитическое сожжение окиси углерода до двуокиси углерода.
2. Поглотительный сосуд-змеевик (6) — наиболее ответственная деталь аналитической части. Служит для поглощения двуокиси углерода раствором едкого бария.
3. Аспиратор, трехходовой кран и уравнительная склянка (16, 17), служащие для засасывания пробы воздуха в прибор, а также для замера его количества. Аспиратор представляет собой цилиндрический сосуд, в центре которого находится трубка, доходящая почти до дна нижней узкой части аспиратора. Тубус аспиратора соединяется посредством резиновой трубки с уравнительной склянкой, заполненной водой. Ниже тубуса в суженной части аспиратора находится слой ртути, препятствующий попаданию воды в прибор при вытеснении воздуха из аспиратора. В верхней, суженной, части аспиратора имеется отвод для удаления проанализированного воздуха из прибора через ртутный затвор.
4. Две микробюретки для титрования растворов едкого бария и соляной кислоты (13, 14) емкостью 4,5 мл с делениями 0,01 мл. В каждую микробюретку напаян сверху полый стеклянный шар, имеющий два отвода. Один отвод шара служит для соединения бюретки с атмосферой через защитный барботер и четырехходовой кран. Другой отвод шара соединен со сливной склянкой и служит для сливания в нее избыточной жидкости.
5. Сливная склянка (12) служит для сливания отработанного раствора из поглотительного сосуда и избытка раствора из микробюреток через трехходовой кран (10).
6. Краны трехходовые (7), позволяющие спускать растворы Ba(OH)2 и HCl в поглотительный сосуд (6), а также для заполнения бюреток растворами. Все краны смазывают полувакуумной смазкой.
7. Четырехходовой кран (11), к которому присоединяется резиновый баллон (19). С помощью резинового баллона создается разрежение в приборе.
8. Для склянки (15) с титрованными растворами. Пробку на склянке с едким барием герметизируют парафином или замазкой.
9. Трансформатор на 2 — 6 В (на рисунке не помечен).
10. Пипетка газовая емкостью 500 мл или бутыль с сифоном объемом не менее 1 л.
11. Защитные барботеры (20, 21).
12. Ртутный затвор (22).
Реактивы
1. Ртуть.
2. Раствор для поглощения паров ртути. Смешивают 30 г йодистого калия и 2,5 г кристаллического йода. В смесь постепенно вливают 1 л дистиллированной воды.
Растворы соляной кислоты, гидрата окиси бария, едкого кали, хлористого натрия и др. готовят так, как указано при определении окиси углерода с йодноватым ангидридом.
Отбор пробы
а) При определении максимально разовой концентрации отбирают две пробы в газовые пипетки емкостью 400 — 500 мл, наполненные 26% раствором поваренной соли. В месте отбора раствор выпускают из пипетки в течение 5 — 10 мин. и таким образом заполняют их исследуемым воздухом. По окончании отбора закрывают зажимы на пипетке, а в концы резиновых трубок вставляют стеклянные палочки.
б) Для определения среднесуточной концентрации отбирают пробу в течение 24 ч в аспиратор емкостью 3 л, наполненный 26% раствором поваренной соли. Скорость отбора 0,1 л/ч. Через 24 ч в верхней бутыли аспиратора должно остаться раствора около 0,5 л.
Подготовка прибора для производства анализа
Для приведения прибора в рабочее состояние необходимо сделать следующие операции:
1. Заполнить бюретки растворами, для чего создают в них разрежение резиновым баллоном путем соединения четырехходового крана с внешним воздухом. В таком положении воздух из баллона выжимают, сжимая его рукой. Не разжимая руку, закрывают кран, соединяют баллон с бюретками и сливной склянкой. В бюретках и сливной склянке при разжимании баллона создается разрежение. Трехходовые краны провертывают таким образом, чтобы бюретки соединились со склянками, наполненными титрованными растворами. При этом бюретки заполняются растворами.
2. Заполнить поглотительный сосуд-змеевик в токе воздуха титрованными растворами. Аспиратор заполняют водой, из аспиратора медленно сливают воду в уравнивательную склянку. Вращая четырехходовой кран, создают в бюретках атмосферное давление. Краны (7) поочередно поворачивают в сторону поглотительного сосуда, соединяя его с бюретками и заполняя их то одним титрованным раствором, то другим, так, чтобы верхние капилляры сосуда погрузились в раствор. Создавая разрежение в бюретках с помощью баллончика (19) и четырехходового крана, засасывают из поглотительного сосуда по капиллярам раствор из бюретки. Если в процессе работы образуются пузырьки в капиллярах и отводах, то их вытесняют так же. По удалении пузырьков воздуха жидкость из поглотительного сосуда выливают в сливную склянку в токе воздуха. Это делают с помощью баллона и четырехходового крана, открывая кран у поглотительного сосуда в сторону сливной склянки, при этом жидкость выливается.
Чтобы вылить жидкость из сливной склянки, следует баллон наполнить воздухом, снять заглушку с резиновой трубки на сливной склянке, сжать баллон рукой, и жидкость из склянки выльется.
После этого необходимо произвести проверку прибора на герметичность. Для этого в поглотительный сосуд спускают растворы едкого бария и соляной кислоты, закрывают заглушкой входное отверстие прибора, наполняют аспиратор водой, спускают уравнительную склянку в нижнее положение, открывают кран на полный ход и включают колонки для сожжения. В таком положении прибор оставляют на 5 мин. Если прибор герметичен, в отдельных его частях не наблюдается движения жидкости. Если прибор не герметичен, то следует тщательно осмотреть его, чтобы установить, в какой части имеется негерметичность. В случае обнаружения негерметичности следует сменить резиновые трубки и снова проверить прибор на герметичность. После этой операции прибор готов для проведения анализа.
Контрольный анализ
Для определения титра раствора едкого бария включают колонку (1) в очистительной системе. Заполняют аспиратор водой, опускают уравнительную склянку вниз, заполняют бюретки растворами, создавая разрежение в бюретках, как описано выше. Затем выравнивают давление в бюретках с помощью четырехходового крана, наливают в поглотительный сосуд 5 — 6 мл раствора едкого бария (точно) в зависимости от размера барботера и пропускают 500 мл воздуха. Не прекращая тока воздуха, проводят титрование раствора едкого бария 0,01 н. раствором соляной кислоты до обесцвечивания раствора.
На контрольный анализ и анализ пробы берут одинаковое количество раствора едкого бария. Контрольный анализ проводят несколько раз до получения совпадающих результатов.
Ход анализа
В поглотительный сосуд наливают 4 — 4,5 мл раствора едкого бария в токе воздуха при включенной колонке в очистительной системе. Присоединяют газовую пипетку с пробой к прибору таким образом, чтобы верхний конец пипетки, закрытый зажимом, был соединен с колонкой предварительного сожжения, а нижний, тоже закрытый зажимом, — с уравнительным сосудом, наполненным водой. Открывая нижний зажим пипетки, создают давление в ней.
Аспиратор заполняют водой до метки «0». Уравнительную склянку аспиратора опускают вниз. Выключают колонку в очистительной системе и включают колонку в аналитической системе. Верхний зажим пипетки с пробой открывают и с помощью аспиратора протягивают анализируемый воздух из пипетки через прибор со скоростью 17 — 20 мл/мин. Отсчет анализируемого воздуха ведут по аспиратору прибора.
Рис. 23. Титрометрический газоанализатор-ГТУ, изготовленный
по МРТУ 42-2067-02 (Б), А — вид сбоку; Б — общий вид
Анализируемый воздух поступает в колонку очистительной системы (не включенную в сеть) в барботеры со щелочью для освобождения от углекислоты, затем в хлоркальциевую трубку (4,а) и в трубки 4,б, 4,в для осушки и очистки воздуха от углеводородов. После очистительной системы воздух поступает в колонку (5), где окись углерода сгорает до углекислоты на раскаленной платиновой спирали.
Образовавшаяся углекислота поступает в поглотительный сосуд, наполненный раствором едкого бария. После сжигания пробы пипетку отъединяют, затем пропускают 100 мл «чистого» воздуха для проталкивания анализируемого воздуха в аналитическую часть, выключая колонку в аналитической системе, включая колонку в очистительной системе, и проводят титрование раствора в поглотительном сосуде в токе воздуха. По разности результатов титрования до и после анализа вычисляют содержание окиси углерода. 1 мл 0,01 н. раствора соляной кислоты соответствует 0,14 мг окиси углерода (полученную величину умножают на 0,14).
Расчет анализа см. выше.
Примечание. Газоанализатор титрометрический: изготавливается по МРТУ 42-2067-62. Изготовитель — завод «Лаборприбор» (рис. 23).
БЕНЗИН
Смесь углеводородов ряда метана, непредельных, ароматических углеводородов, нафтенов.
Существуют нефтяные, сланцевые, синтетические и другие бензины.
Нефтяные бензины — продукты, получаемые в процессе перегонки нефти.
Сланцевый бензин — продукт пиролиза горючих сланцев.
Синтетические бензины — продукты гидрирования окиси углерода, ацетилена и др. и дальнейшей полимеризации.
Бензины представляют собой легко испаряющиеся жидкости, без цвета или с желтоватым оттенком.
Химический состав различных бензинов отличается и зависит от способов их получения. В состав сланцевых бензинов входят сернистые органические соединения.
Бензины обладают наркотическим действием, оказывают влияние на центральную нервную систему. Вызывают головную боль, раздражение слизистых оболочек, дерматиты.
Предельно допустимые концентрации (мг/м3):
|
Максимальная разовая |
Среднесуточная |
|
|
Бензин (нефтяной, малосернистый в пересчете на углерод) |
5 |
1,5 |
|
Бензин сланцевый (в пересчете на углерод) |
0,05 |
0,05 |
ОПРЕДЕЛЕНИЕ БЕНЗИНА (СУММЫ УГЛЕВОДОРОДОВ)
НА ГАЗОАНАЛИЗАТОРЕ [4]
Принцип определения
При каталитическом сожжении углеводородов (бензина, спирта и других легких углеводородов) происходит образование углекислого газа, который поглощается раствором гидрата окиси бария. Его избыток оттитровывается раствором соляной кислоты. Концентрацию суммы углеводородов пересчитывают на углерод (C). Мешают определению органические соединения, которые поглощаются щелочью, кислотой или реагируют с ними (альдегиды, кетоны, фенолы, сложные эфиры), а также галогеноорганические соединения.
Чувствительность определения в пересчете на углерод при титровании 0,01 н. раствором соляной кислоты 0,0006 мг (0,6 мкг), при титровании 0,005 н. раствором соляной кислоты 0,0003 мг (0,3 мкг).
Метан мешает определению.
Прибор, реактивы, отбор проб, подготовка прибора к анализу и определение титра гидрата окиси бария описаны при определении окиси углерода на газоанализаторе (см. выше). При анализе пробы на содержание бензина V-образные трубки, наполненные силикагелем и пемзой, пропитанной серной кислотой (поглощающие углеводороды), отключают от прибора.
ОПРЕДЕЛЕНИЕ БЕНЗИНА (СУММЫ УГЛЕВОДОРОДОВ)
В ПРИСУТСТВИИ ОКИСИ УГЛЕРОДА НА ГАЗОАНАЛИЗАТОРЕ
Принцип определения
Метод основан на каталитическом сожжении бензина и окиси углерода до CO2, поглощении двуокиси углерода раствором барита и обратном титровании его избытка раствором соляной кислоты; в той же пробе по этому принципу определяется окись углерода, а затем до разности находят концентрацию бензина в пересчете на углерод.
Чувствительность определения в пересчете на углерод 0,0006 мг (0,6 мкг). Мешает определению присутствие других органических веществ. Аппаратура, реактивы, отбор пробы, подготовка прибора к анализу, определение титра едкого барита см. «Определение окиси углерода на газоанализаторе» выше.
Контрольный анализ
Через прибор в поглотительный сосуд с едким баритом при включенных колонках протягивают 500 мл «чистого» воздуха, титруют раствор барита раствором HCl, отмечая количество миллилитров HCl, пошедшее на обратное титрование гидроокиси бария (а).
Анализ пробы
Сначала определяют общее содержание углеводородов и окиси углерода в исследуемом воздухе. При этом колонки очистительной системы (колонки с пемзой, пропитанной H2SO4 и силикагелем) отсоединяют. Силикагель используют марки СК и СМ, размер зерен 1 — 2 мм. Его предварительно активируют при 250°C. Спускают из бюретки точно такое же количество раствора едкого барита, какое было взято при проведении контрольного анализа.
Короткий конец бутыли с пробой присоединяют к прибору, другой конец сифона соединяют со склянкой, наполненной 26% раствором хлористого натрия. Открывают зажимы сифона бутыли сначала длинной трубки, проверяя герметичность бутыли (если бутыль герметична, то через короткое время прекращается поступление раствора в бутыль). Затем открывают зажим короткой трубки, и солевой раствор, поступая в бутыль с пробой воздуха, вытесняет последний в прибор.
Объем пропускаемого воздуха учитывают по аспиратору прибора. Выключают колонку предварительного сожжения и оставляют включенной колонку аналитической части, с помощью аспиратора и уравнительной склянки протягивают исследуемый воздух со скоростью 17 — 20 мл/мин. Образующаяся при сожжении бензина и окиси углерода двуокись углерода поступает в поглотительный сосуд с раствором барита. После вытеснения 500 мл исследуемого воздуха пробы из бутылки последнюю отсоединяют от прибора и присоединяют к прибору очистительную систему (колонки, заполненные пемзой, пропитанной серной кислотой и силикагелем), пропуская через нее 100 мл «чистого» воздуха для проталкивания остатка анализируемого воздуха в аналитическую часть. Затем включают колонку предварительного сожжения и пропускают еще 100 мл «чистого» воздуха, затем производится титрование избытка барита раствором HCl с отметкой количества соляной кислоты в миллилитрах.
Определение окиси углерода
Определение окиси углерода в пробе производится так же, как было указано выше, с той лишь разницей, что анализируемый воздух до поступления в барботеры предварительно очищают от бензина пропусканием через очистительную систему (колонки — одну, заполненную пемзой, пропитанной серной кислотой, и другую, заполненную силикагелем), отмечают количество соляной кислоты в миллилитрах, пошедшее на титрование избытка барита. Концентрацию бензина в пробе воздуха в пересчете на углерод (мг/м3) рассчитывают по формуле:
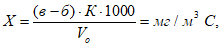
где: в — количество соляной кислоты (мл), пошедшее на титрование избытка раствора барита после поглощения CO2, образовавшегося при сожжении окиси углерода и углеводородов пробы воздуха; б — количество соляной кислоты (мл), пошедшее на титрование избытка раствора барита после поглощения CO2, образовавшегося при сожжении окиси углерода пробы воздуха; K = 0,06 — количество C (мг), соответствующее 1 мл 0,01 н. раствора HCl; V0 — количество исследуемого воздуха (л), приведенное к нормальным условиям (см. выше).
В случае необходимости вычислить и концентрацию окиси углерода в исследуемой пробе (в миллиграммах на 1 м3 воздуха) (X), ее рассчитывают по следующей формуле:
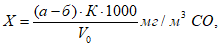
где: а — количество миллилитров HCl, пошедшее на титрование избытка раствора барита после протягивания «чистого» воздуха, при контрольном
анализе; б — количество HCl в 1 мл, пошедшее на титрование избытка раствора барита после поглощения CO2, полученного при сожжении окиси углерода из пробы воздуха; K = 0,14 — количество CO в миллиграммах, соответствующее 1 мл 0,01 н. раствора HCl; V0 — количество исследуемого воздуха пробы в литрах, приведенного к нормальным условиям (см. выше).
ЦИАНИСТЫЙ ВОДОРОД (СИНИЛЬНАЯ КИСЛОТА)
Бесцветная, легко подвижная, весьма летучая жидкость с запахом горького миндаля. Хорошо растворима в воде, органических растворителях. Плотность паров по отношению к воздуху 0,93. Температура кипения 25,5 °C, температура замерзания — 14 °C. Упругость паров 647,9 мм рт. ст. при 21,4 °C. Цианистый водород относится к сильно токсическим соединениям, вызывает параличи дыхательных путей, проникает через кожные покровы. Среднесуточная предельно допустимая концентрация 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИАНИСТОГО ВОДОРОДА
ПО ОБРАЗОВАНИЮ ПОЛИМЕТИНОВОГО КРАСИТЕЛЯ [1]
Принцип определения
При взаимодействии цианистого водорода и пиридина с анилином образуется дианилид глютаконового альдегида, относящийся к классу полиметиновых красителей.
Чувствительность определения 0,1 мкг в анализируемом объеме раствора.
Мешают определению хлор, бром, влияние их устраняют поглощением в индикаторную трубку.
Реактивы
1. Поглотительный раствор — 0,1 н. раствор едкого натрия.
2. Исходный стандартный раствор из фиксанала 0,1 н. раствора роданида аммония, 1 мл 0,1 н. раствора которого соответствует 5,81 мг иона SCN или 2,7 мг CN.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят перед употреблением соответствующим разбавлением водой.
3. Соляная кислота, 0,1 н. раствор.
4. Едкий натр, 0,01 н. раствор.
5. Бромная вода. Смешивают 0,5 мл брома с 50 мл воды.
6. Пиридин. Очищают кипячением в течение часа с обратным холодильником над кристаллической щелочью. Затем, добавив кристаллическую щелочь, перегоняют. Отбирают фракцию, кипящую при 114 — 116°. Хранят в темной закрытой склянке.
7. Анилин. Перегоняют при 184,5 °C.
8. Уксусная кислота, ледяная, х.ч.
9. Индикаторная вата. Гигроскопическую вату промывают горячим спиртом, высушивают при 80 — 90 °C. В раствор, состоящий из 40 г йодида калия и 100 мл воды, погружают на 20 мин. 10 г ваты. Вату отжимают между фильтровальной бумагой и высушивают при 80 °C. Хранят в склянке из темного стекла.
10. Составной реактив готовят смешением 10 мл пиридина, 5 мл ледяной уксусной кислоты и 1 мл анилина.
11. Гидразинсульфат, 0,5% раствор.
Отбор пробы
Воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 2 мл поглотительного раствора. При наличии хрома, брома в воздухе перед поглотительными приборами включают индикаторную трубку с ватой. Протягивают не менее 80 л исследуемого воздуха.
Ход анализа
Из каждого поглотительного прибора 1 мл пробы помещают в пробирки, прибавляют по 1 мл 0,1 н. соляной кислоты и 0,1 мл бромной воды и по каплям 0,5% раствора гидразинсульфата до обесцвечивания. Перемешивают и вносят по 1 мл составного реактива и доводят объем до 4 мл водой. Через 15 мин. фотометрируют в кюветах с толщиной слоя 1 см при длине волны 485 нм по сравнению с контролем, который готовят одновременно с пробой. Содержание синильной кислоты определяют по предварительно построенному калибровочному графику. Готовят калибровочный график (табл. 36).
Таблица 36
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание синильной кислоты, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
Все пробирки шкалы стандартов и пробу обрабатывают, аналогично измеряют оптические плотности и строят график. Шкала стандартов может быть использована для визуального определения.
Расчет см. выше.
ЛИТЕРАТУРА
1. Беляков А.А., Мельникова Л.В. Определение синильной кислоты. В кн.: Определение вредных веществ в воздухе производственных помещений. Волго-вятское книжное издательство. Горький, 1970.
2. Методы определения окиси углерода в воздухе. В сб.: Технические условия на методы определения вредных веществ в воздухе. М., 1964, в. III.
3. Сендерихина Д.П. Определение окиси углерода на титрометрическом газоанализаторе. В сб.: Информационно-методические материалы. Методы гигиенических исследований. Московский научно-исследовательский институт санитарии и гигиены им. Ф.Ф. Эрисмана. М., 1955, в. 5.
4. Сендерихина Д.П. Определение бензина в присутствии окиси углерода на газоанализаторе. В сб.: Методы гигиенических исследований. Информационно-методические материалы. Московский научно-исследовательский институт санитарии и гигиены им. Ф.Ф. Эрисмана. М., 1955, в. 5.
Глава VIII
УГЛЕВОДОРОДЫ АРОМАТИЧЕСКОГО РЯДА
БЕНЗОЛ
Бесцветная прозрачная жидкость с характерным запахом, плотность при 20° 0,879. Температура плавления 5,53 °C; температура кипения 80,1°. Бензол хорошо растворим в эфире, спирте, хлороформе и других органических растворителях; он является хорошим растворителем жиров, смол, масел, ряда других органических веществ; растворяет серу, фосфор и йод.
Высокие концентрации бензола оказывают влияние на центральную нервную систему, а также вызывают изменения крови и кроветворных органов.
Бензол довольно сильно раздражает кожу.
Предельно допустимые концентрации: максимальная разовая 1,5 мг/м3; среднесуточная 0,80 мг/м3.
ОПРЕДЕЛЕНИЕ БЕНЗОЛА НИТРОВАНИЕМ [1]
Принцип определения
При нитровании бензола образуется динитробензол, который в щелочной среде в присутствии кетона дает соединение, окрашенное в красно-фиолетовый цвет.
Чувствительность определения 1 мкг в анализируемом объеме раствора. Другие ароматические углеводороды мешают определению.
Реактивы
1. Поглотительный раствор — нитрационная смесь: 10 г нитрата аммония, высушенного при 80 °C, растворяют в 100 мл серной кислоты, уд. вес 1,84.
2. Стандартный раствор бензола: исходный раствор готовят в мерной колбе емкостью 50 мл, в которой помещено 1 — 2 мл нитрационной смеси. Колбу взвешивают и вносят в нее 2 — 3 капли бензола и вновь взвешивают. По разности двух взвешиваний устанавливают количество бензола в колбе. Раствор встряхивают и колбу помещают в кипящую водяную баню на 40 мин. По истечении времени раствор охлаждают и постепенно, осторожно в колбу вливают воду при помешивании раствора. Колбу в это время помещают в холодную водяную баню. Раствор доводят до метки после того, как температура его сравнится с комнатной.
Высчитывают содержание бензола в 1 мл раствора. Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 5 мг бензола. Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
3. Метилэтилкетон (бутанон) бесцветный, свежеперегнанный, точка кипения 74 — 80 °C. Бутанон, бывший в употреблении, можно применять вновь, предварительно отделив от щелочи в делительной воронке, промыв водой, осушив хлоридом кальция, дважды перегнав его. Полученный бутанон не должен давать окраску со щелочью.
4. Едкий натр или едкое кали, 20% раствор.
5. Серная кислота, уд. вес 1,84.
6. Бензол, свежеперегнанный (температура кипения 80,1 °C).
7. Нитрат аммония, высушенный при 80 °C.
8. Аммиак, 25% раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1 (малая модель), содержащий 2 мл поглотительного раствора. Продолжительность отбора 20 — 40 мин.
б) Для определения среднесуточной концентрации воздух в течение суток протягивают через поглотительный прибор непрерывно, со скоростью 0,1 л/мин.
Допустимо отбор проб проводить 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствора его сливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 6 — 8 мл. Воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения пробу нейтрализуют 25% раствором аммиака по лакмусу. Нейтрализацию проводят из бюретки с краном при охлаждении.
Затем пробу переводят в делительную воронку емкостью 25 мл, добавляют 4 мл бутанона и раствор встряхивают в течение 5 мин. Раствору дают расслоиться и водный нижний слой выливают через кран воронки, а верхний бутаноновый переливают через верх воронки в пробирку, объем раствора доводят до 4 мл бутаноном.
В пробирку с пробой наливают 1 мл 20% раствора едкого натра, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 20 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 574 нм. Содержание бензола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов из стандартного бутанонового раствора. Для этого 2 мл рабочего стандартного раствора вливают осторожно в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и приливают к нему 10 мл бутанона. Растворы встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Затем нижний водный слой сливают через кран воронки, а верхний бутаноновый слой переливают через верхний край воронки в мерную колбу емкостью 10 мл. Объем раствора доводят бутаноном до метки, в 1 мл этого раствора содержится 20 мкг бензола. Из этого раствора готовят шкалу стандартов (табл. 37).
Таблица 37
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
|
Бутанон, мл |
4 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
|
|
Содержание бензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой. В первой пробирке может содержаться 0,5 мкг и далее, как указано в табл. 37.
Расчет см. выше.
ТОЛУОЛ
Бесцветная жидкость с характерным запахом. Плотность 0,867 при 20 °C, температура кипения 110,6 °C, смесь паров толуола с воздухом взрывоопасна. Толуол растворим в ряде органических растворителей.
Толуол обладает наркотическим действием, действует на нервную систему, кроветворные органы и др.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,6 мг/м3.
ОПРЕДЕЛЕНИЕ ТОЛУОЛА НИТРОВАНИЕМ [2]
Принцип определения
При нитровании толуола образуется тринитротолуол, который в щелочной среде в присутствии кетона дает соединение, окрашенное в оранжево-розовый цвет.
Чувствительность определения — 2 мкг в определяемом объеме раствора. Другие ароматические углеводороды мешают определению.
Реактивы
1. Стандартный раствор толуола готовят из перегнанного. Исходный раствор получают в мерной колбе емкостью 50 мл, в которую внесено 1 — 2 мл нитрационной смеси. Колбу взвешивают, вносят 2 — 3 капли толуола и снова взвешивают. По разности двух взвешиваний устанавливают количество толуола. Раствор встряхивают и колбу помещают на 30 мин. в кипящую водяную баню. Раствор охлаждают и в него осторожно вливают воду. Колба в это время должна быть помещена в холодную водяную баню. Высчитывают содержание толуола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разведением исходного раствора. Объем доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3. Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 30 мин. По охлаждении раствор переливают в коническую колбу емкостью 50 мл, а поглотительный прибор ополаскивают несколько раз водой (5 — 6 мл).
Воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении в присутствии лакмусовой бумаги.
Затем пробу переводят в делительную воронку на 25 — 30 мин. с хорошо притертой пробкой, добавляют 2 мл бутанона и раствор встряхивают в течение 5 мин., дают расслоиться и водный нижний слой сливают через кран воронки, а верхний бутаноновый — через верх воронки в пробирку. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл 20% раствора щелочи и содержимое пробирки встряхивают в течение нескольких минут. Через 15 — 20 мин. измеряют оптическую плотность раствора в кювете с толщиной слоя 0,5 — 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят с помощью фотоэлектроколориметра при длине волны 453 нм.
Содержание толуола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую предварительно вливают 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса. Охлажденный нейтральный раствор выливают в делительную воронку и в нее вливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного расслоения раствора. Нижний водный слой сливают через кран воронки, а верхний бутаноновый слой переливают через верхний край воронки в мерную колбу емкостью 10 мл. Объем раствора доводят до метки бутаноном. 1 мл этого раствора содержит 10 мкг толуола. Из этого раствора готовят шкалу стандартов (табл. 38).
Таблица 38
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Тутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание толуола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
КСИЛОЛ
|
C6H4(CH3)2 |
Мол. вес 106,17 |
Бесцветная жидкость. Ксилол может быть в виде трех изомеров: ортоксилол — температура кипения 144,4 °C, плотность при 20 °C 0,881; метаксилол — температура кипения 139,1 °C, плотность при 20° 0,864; параксилол, температура кипения 138,35 °C, плотность при 20 °C 0,861.
Пары ксилола тяжелее воздуха в 3,7 раза, взрывоопасны. Технический ксилол является смесью трех изомеров. Растворимость в воде при 22 °C 0,013%.
Ксилол — наркотик, действие его на организм человека сходно с бензолом и толуолом, действуют они на кроветворные органы.
Предельно допустимая концентрация: максимальная разовая и среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ КСИЛОЛА НИТРОВАНИЕМ [3]
Принцип определения
При нитровании ксилола образуется нитросоединение ксилола, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в серо-зеленый цвет.
Чувствительность определения 2 мкг в определяемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор готовят из свежеперегнанного ксилола при 138 — 145 °C. Исходный раствор получают в мерной колбе емкостью 50 мл, в которой помещено 1 — 5 мл нитрационной смеси. Колбу взвешивают и в нее вносят 2 — 3 капли ксилола. По разности двух взвешиваний устанавливают количество ксилола в 1 мл. Колбу помещают в кипящую водяную баню на 30 мин. По охлаждении в колбу вносят малыми порциями дистиллированную воду до метки.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл соответствующим разбавлением исходного стандартного раствора.
Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 30 мин. По охлаждении раствора его переливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 5 — 6 мл. Воду приливают к пробе осторожно, избегая сильного разогревания.
После охлаждения пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении раствора в присутствии лакмуса. Затем пробу переводят в делительную воронку емкостью 25 — 30 мл с хорошо притертой пробкой, добавляют к пробе 2,2 мл бутанона и раствор встряхивают в течение 5 мин. Раствору дают расслоиться и водный нижний слой сливают через кран воронки, а верхний, бутаноновый, переливают через верх воронки в пробирку. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл щелочи и содержимое пробирки встряхивают в течение нескольких минут. Через 15 — 20 мин. измеряют оптическую плотность в кювете толщиной слоя 0,5 — 1 см по сравнению с контролем, который готовят одновременно и аналогично. Определение проводят с помощью фотоэлектроколориметра при длине волны 508 нм.
Содержание ксилола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую было влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и в нее вливают 10 мл бутанона. Растворы встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний водяной слой сливают через кран, а верхний бутаноновый через верх воронки сливают в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Раствор доводят до метки бутаноном, в 1 мл этого раствора содержится 10 мкг ксилола.
Из этого раствора готовят шкалу стандартов (табл. 39).
Таблица 39
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Бутанон, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
|
Содержание ксилола, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ИЗОПРОПИЛБЕНЗОЛ
|
C6H5CH(CH3)2 |
Мол. вес 120,19 |
Бесцветная жидкость, температура кипения 152,39 °C, плотность 0,86 при 20 °C. Хорошо растворим в органических растворителях. В воде не растворим.
Изопропилбензол токсичен.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,014 мг/м3.
ОПРЕДЕЛЕНИЕ ИЗОПРОПИЛБЕНЗОЛА НИТРОВАНИЕМ [4]
Принцип определения
При взаимодействии изопропилбензола с нитрационной смесью образуется полинитросоединение. После извлечения нитросоединения эфиром, растворения сухого остатка в этаноле в присутствии щелочи раствор окрашивается в оранжевый цвет.
Чувствительность определения — 1 мкг изопропилбензола в определяемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор готовят из свежеперегнанного изопропилбензола. Исходный стандартный раствор готовят в мерной колбе емкостью 25 — 50 мл, в которую вносят 5 — 10 мл нитрационной смеси. Колбу взвешивают, вносят 1 — 2 капли изопропилбензола и вновь взвешивают.
По разности взвешиваний устанавливают количество изопропилбензола. Раствор встряхивают и помещают в кипящую водяную баню на 1 ч. После охлаждения раствора его доводят до метки нитрационной смесью. Рассчитывают содержание изопропилбензола в 1 мл.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разбавлением исходного нитрационной смесью.
Приготовление других реактивов и отбор пробы см. «Определение бензола нитрованием» выше.
Для установления содержания изопропилбензола в пределах предельно допустимой концентрации необходимо протянуть не менее 70 л исследуемого воздуха.
Ход анализа
а) Извлечение нитросоединения бутаноном
Поглотительный прибор с пробой помещают в кипящую водяную баню на 1 ч. По охлаждении раствора его переливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 5 — 6 мл, воду приливают к пробе осторожно, избегая сильного разогревания раствора.
После охлаждения раствора пробу нейтрализуют 25% раствором аммиака. Нейтрализацию проводят из бюретки с краном при охлаждении в присутствии лакмуса. Затем пробу переводят в делительную воронку, добавляют 2,2 мл бутанона и раствор встряхивают в течение 5 мин. Жидкость оставляют в покое для расслоения. Нижний водный слой сливают через кран воронки, а верхний, бутаноновый, переливают в пробирку через верх воронки. Объем бутанонового раствора доводят до 2 мл бутаноном. В пробирку наливают 0,75 мл щелочи, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 30 мин. сравнивают интенсивность окраски пробы со шкалой стандартов, которую готовят одновременно с пробой.
Шкалу стандартов готовят из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку, в нее вливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний, водный, раствор сливают через кран воронки, а верхний, бутаноновый, — в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Объем раствора доводят до метки бутаноном. Получают стандартный бутаноновый раствор с содержанием 10 мкг/мл, из него готовят шкалу стандартов (табл. 40). Все пробирки шкалы обрабатывают аналогично и одновременно с пробой.
Таблица 40
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
|
Содержание изопропилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
б) Извлечение нитросоединения эфиром
Поглотительный прибор с пробой помещают на 1 ч в кипящую водяную баню, затем охлаждают и переводят в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают водой несколько раз, на что расходуют 5 — 6 мл воды. Промывную воду приливают к пробе осторожно, избегая сильного разогревания раствора.
По охлаждении содержимое колбы переливают в делительную воронку и добавляют 5 мл эфира. Раствор встряхивают в течение 5 мин. и оставляют в покое для полного разделения слоев. Затем водный, нижний, слой сливают через кран воронки, а верхний оставляют в воронке и к нему приливают 5 мл воды. Раствор вновь встряхивают. После разделения растворов нижний слой, водный, выливают через кран воронки, а верхний, эфирный, переливают через верх воронки в фарфоровую чашку и испаряют эфир.
Сухой остаток постепенно растворяют в 2 мл этанола и раствор переносят в пробирку с меткой 2 мл. В пробирку вливают 0,5 мл 5% раствора спиртовой щелочи и встряхивают в течение нескольких минут.
Одновременно готовят шкалу стандартов (табл. 41).
Таблица 41
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
0,9 |
|
Этанол, мл |
2 |
1,9 |
1,8 |
1,6 |
1,6 |
1,2 |
1,0 |
|
Содержание изопропилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
При извлечении нитросоединения, как эфиром, так и бутаноном, определение проводят визуально через 25 — 30 мин. после добавления щелочи.
Расчет см. выше.
ЭТИЛБЕНЗОЛ
Прозрачная бесцветная жидкость. Плотность при 20 °C 0,86, температура кипения 136,15 °C. Этилбензол плохо растворим в воде, в 100 мл воды при 15 °C растворяется 4,014 г, в спирте и эфире растворим хорошо. Пары этилбензола вызывают слезотечение и боль в глазах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ЭТИЛБЕНЗОЛА НИТРОВАНИЕМ [6]
Принцип определения
При взаимодействии этилбензола с нитрационной смесью образуется тринитроэтилбензол, который в щелочной среде в присутствии кетона дает окрашенное соединение.
Чувствительность определения 1 мкг этилбензола в анализируемом объеме раствора. Другие ароматические соединения мешают определению.
Реактивы
1. Стандартный исходный раствор этилбензола.
В мерную колбу емкостью 25 мл вносят 10 мл нитрационной смеси. Колбу взвешивают, в нее вливают 1 — 2 капли этилбензола и снова взвешивают. По разности двух взвешиваний определяют количество этилбензола в колбе. Колбу помещают на 30 мин. в кипящую водяную баню и по охлаждении раствора доводят его объем до метки нитрационной смесью и хорошо перемешивают.
Высчитывают содержание этилбензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 25 мл из исходного стандартного раствора. Для этого берут такое количество этого раствора, в котором содержится 250 мкг этилбензола. Объем жидкости в колбе доводят до метки нитрационной смесью.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Для определения этилбензола в пределах максимальной разовой концентрации необходимо протянуть не менее 50 мл исследуемого воздуха.
Ход анализа
Поглотительный прибор с пробой помещают на 30 мин. в кипящую водяную баню. По истечении этого времени пробу охлаждают и осторожно переливают в делительную воронку, в которую предварительно наливают 6 — 8 мл воды. При вливании необходимо избегать сильного разогревания раствора. Поглотительный прибор промывают дважды по 2 мл воды. Воду каждый раз осторожно вливают в делительную воронку.
По охлаждении раствора в воронке в нее вливают 5 мл эфира. Раствор встряхивают в течение 5 мин. и оставляют в покое до полного расслоения. Нижний, водный, слой сливают через кран, верхний оставляют в воронке, к нему приливают 5 мл воды, насыщенной эфиром, и взбалтывают для удаления из эфирного слоя следов серной кислоты. После расслоения растворы разделяют — эфирный выливают в чашку и испаряют эфир почти досуха при комнатной температуре в вытяжном шкафу вдали от огня.
Остаток эфирного раствора переводят в колориметрическую пробирку с притертой пробкой и объем раствора доводят эфиром до 1 мл; предварительно этим эфиром ополаскивают фарфоровую чашку. Затем фарфоровую чашку ополаскивают 1 мл этанола и раствор выливают в ту же пробирку. Объем доводят до 2 мл. Содержимое пробирки перемешивают, вливают 0,1 мл 5% спиртового раствора щелочи и вновь встряхивают. Через 10 — 15 мин. интенсивность окраски пробы сравнивают с окраской шкалы стандартов, которую готовят одновременно с пробой (табл. 42).
Таблица 42
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Нитросмесь, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание этилбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Каждую пробирку шкалы обрабатывают аналогично пробе.
Расчет см. выше.
НИТРОБЕНЗОЛ
Бесцветная жидкость с запахом горького миндаля, температура кипения 210,9 °C, температура плавления 5,7 °C. Нитробензол слабо растворим в воде, хорошо растворяется в спирте, эфире; перегоняется с водяным паром.
Нитробензол токсичен, отравление возможно при вдыхании паров и через кожу; он является кровяным ядом.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ НИТРОБЕНЗОЛА НИТРОВАНИЕМ [5]
Принцип определения
При взаимодействии нитробензола с нитрационной смесью образуется динитросоединение, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в красно-фиолетовый цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор нитробензола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую влито 12 мл нитрационной смеси. Колбу взвешивают и вносят в нее 2 — 3 капли нитробензола и вновь взвешивают. По разности двух взвешиваний устанавливают количество нитробензола в колбе. Раствор встряхивают и помещают в кипящую водяную баню на 40 мин. По истечении этого времени раствор охлаждают и постепенно, осторожно в колбу вливают воду при помешивании раствора. Колбу в это время помещают в холодную водяную баню. Раствор доводят до метки после того, как температура его сравняется с комнатной. Высчитывают содержание нитробензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 5000 мкг нитробензола. Раствор доводят до метки нитрационной смесью, разбавленной водой в отношении 1:3.
2. Нитробензол свежеперегнанный, температура кипения 210,9 °C.
Приготовление других реактивов и отбор пробы см. «Определение бензола нитрованием» выше. Для определения нитробензола в пределах предельно допустимой концентрации необходимо протянуть не менее 70 л исследуемого воздуха.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствора его сливают в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз водой, на что расходуют 6 — 8 мл. Воду приливают к пробе осторожно, избегая сильного разогревания.
После охлаждения раствора его нейтрализуют 25% раствором аммиака по лакмусу. Нейтрализацию проводят из бюретки с краном при охлаждении раствора. Затем пробу переводят, в делительную воронку емкостью 25 — 30 мл и добавляют 2,2 мл бутанона. Раствор встряхивают в течение 5 — 10 мин. и дают расслоиться. Водный нижний слой сливают через кран воронки, верхний бутаноновый слой переливают через верхний край воронки в пробирку и объем раствора доводят до 2 мл бутаноном.
В раствор вливают 0,75 мл 20% раствора щелочи, содержимое пробирки встряхивают в течение нескольких минут и через 15 — 20 мин. измеряют оптическую плотность в кювете с толщиной слоя 0,5 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 536 нм.
Содержание нитробензола в анализируемом объеме определяют по предварительно поставленному калибровочному графику. Для построения графика готовят шкалу стандартов из стандартного бутанонового раствора, для получения его берут 1 мл рабочего стандартного раствора, вливают осторожно в коническую колбу емкостью 100 мл, в которую предварительно влито 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор переливают в делительную воронку, к нему приливают 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Затем нижний водный слой удаляют через кран воронки, а верхний бутаноновый — через верх воронки, вливают в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Этот раствор содержит нитробензола 10 мкг/мл.
Из этого раствора готовят шкалу стандартов (табл. 43).
Таблица 43
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание нитробензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ДИНИЛ
Динил — это смесь 26,5% раствора дифенила и 73,5% раствора дифинилового эфира. Жидкость с характерным резким запахом, температура кипения 258 °C.
Динил является токсичным веществом, он вызывает изменение в почках, печени и в других органах.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ДИНИЛА НИТРОВАНИЕМ [8]
Принцип определения
Метод основан на определении окрашенного продукта реакции, образовавшегося при взаимодействии нитропроизводного динила с ацетоном и сульфидом натрия.
Чувствительность определения — 0,25 мкг в анализируемом объеме раствора. Метадинитробензол определению не мешает. Влияние других гомологов бензола устраняют применением хромовой смеси для их окисления в процессе анализа.
Реактивы
1. Поглотительный раствор — нитрационная смесь. 10 г нитрата калия и 0,5 г сернистокислого натрия растворяют в 100 мл серной кислоты уд. веса 1,84.
2. Стандартный раствор динила. Исходный раствор готовят в мерной колбе емкостью 50 мл, в которую наливают 1 — 2 мл нитрационной смеси, колбу взвешивают, вносят 2 — 3 капли динила и снова взвешивают. По разности устанавливают количество динила в колбе и рассчитывают его содержание в 1 мл раствора. Стандартный раствор с содержанием 100 мкг/мл и рабочий стандартный раствор с содержанием 10 мг/мл готовят соответствующим разведением нитросмесью.
3. Толуол.
4. Ацетон.
5. Натрий углекислый, 2% раствор, подкрашенный фенолфталеином до розовой окраски.
6. Натрий сернистый, 1% раствор водный или спиртовой (хранят в закрытой склянке не более месяца).
7. Уксусная кислота, 10% раствор.
8. Калий двухромовокислый, 10% раствор.
9. Фенолфталеин, 0,1% раствор.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористым фильтром, наполненный 2,5 мл нитрационной смеси. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с 5 мл нитрационной смеси в течение суток. Допустимо проводить отбор в один и тот же поглотительный прибор с перерывами в 2 — 4 ч.
Ход анализа
В поглотительный прибор вводят 0,5 мл воды, перемешивают, используя для этого резиновую грушу. Прибавляют 1 мл 10% раствора двухромовокислого калия, перемешивают и помещают на 30 мин. в кипящую водяную баню. При отборе пробы в 5 мл нитрационной смеси в поглотитель вводят 1 мл воды и 2 мл раствора бихромата.
После нагрева в поглотитель вводят 10 мл воды, охлаждают и сливают в делительную воронку. Поглотительный прибор ополаскивают 2 раза по 2 мл воды, сливая ее в воронку. В воронку вводят 2 мл толуола для экстрагирования и интенсивно взбалтывают в течение 2 — 3 мин. После расслоения водный слой отделяют, а экстракт промывают 3 — 5 мл раствора углекислого натрия. При обесцвечивании раствора соды его прибавляют в количестве 1 — 2 мл.
Содовый слой отделяют, экстракт сливают через верх воронки в пробирку. Делительную воронку ополаскивают 0,5 мл толуола, который присоединяют к экстракту. В пробирку добавляют ацетона до 10 мл и 0,05 мл 1% раствора сульфида натрия, перемешивают и через 2 мин. подкисляют 0,3 мл 10% раствора уксусной кислоты. Через 5 — 10 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 450 нм по сравнению с контролем. Контроль готовят одновременно и аналогично пробе. Содержание динила определяют по заранее построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 44).
Таблица 44
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,025 |
0,05 |
0,075 |
0,1 |
0,15 |
0,2 |
|
Нитрационная смесь, мл |
2,5 |
2,475 |
2,450 |
2,425 |
2,4 |
2,35 |
2,25 |
|
Количество динила, мкг |
0 |
0,25 |
0,5 |
0,75 |
1,0 |
1,5 |
2,0 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Расчет см. выше.
СТИРОЛ
|
C6H5·CH=CH2 |
Мол. вес 104,15 |
Бесцветная жидкость с температурой кипения 146 °C, температурой плавления 33 °C. Плотность ее 0,909 (20°); упругость пара 10 мм рт. ст. (33°). Стирол трудно растворим в воде, хорошо растворим в спирте, эфире, метаноле, ацетоне и других органических растворителях. Стирол полимеризуется в полистирол, особенно этот процесс ускоряется с повышением температуры. Стирол обладает наркотическим действием, раздражающим действием на слизистые оболочки, вызывает поражение печени, а также действует на кроветворные органы. Предельно допустимые концентрации стирола: максимальная разовая 0,003 мг/м3, среднесуточная 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ СТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ [7]
Принцип определения
При взаимодействии стирола с ацетатом ртути образуется ртутно-органическое производное, которое выделяют из смеси на бумаге нисходящим способом в системе растворителей н-бутанол — вода — диэтиламин. При орошении хроматограммы раствором дифенилкарбазида производные стирола проявляются в виде фиолетовых зон, Rf = 0,8.
Количественное определение стирола проводят фотометрически по измерению оптической плотности элюата или визуально, сравнивая интенсивность окраски зон локализации пробы и шкалы.
Чувствительность определения 1 мкг стирола в анализируемом объеме раствора. Изопропилбензол, бензол, гидроперекись изопропилбензола, перекись бензоила, малеиновый, фталевый ангидриды, диметиланилин, дивинил, аммиак, дитолилметан, изопрен, хлоропрен не мешают определению.
Аппаратура
1. Теплоэлектровентилятор.
2. Хроматографическая камера, размер 240 на 600 мм.
3. Микропипетки емкостью 0,1 — 0,2 мл с делениями на 0,001 мл.
4. Пробирки высотой 9 см, диаметром 0,8 см с делениями 0,1 — 0,2 — 0,3 мл.
5. Стеклянный распылитель.
Реактивы
1. Поглотительный раствор. К 100 мл этанола добавляют 0,1 г ацетата ртути и 0,5 мл уксусной кислоты.
2. Ацетат ртути, двузамещенная соль.
3. Этиловый спирт, очищенный. К 1 л этанола добавляют 40 г едкого кали, оставляют на 2 ч, перегоняют при 78 °C.
4. Кали едкое, х.ч.
5. н-Бутанол.
6. Диэтиламин.
7. Дифенилкарбазид, 0,1% раствор в этаноле.
8. Хроматографическая бумага «Медленная».
9. Стандартный раствор стирола. Исходный раствор готовят из свежеперегнанного стирола (при 145 °C). В мерную колбу вносят 2 — 3 мл поглотительного раствора, взвешивают. Добавляют 1 — 2 капли стирола и снова взвешивают. Объем 25 мл колбы доводят до метки поглотительным раствором. Рассчитывают содержание стирола в 1 мл. Соответствующим разбавлением поглотительным раствором готовят раствор N 1 с содержанием 100 мкг/мл.
10. Система растворителей. Готовят смесь н-бутанола, воды, диэтиламина, 5:4:1. Смесь помещают в делительную воронку, перемешивают. После разделения слоев нижний используют для смачивания фильтровальной бумаги с целью насыщения хроматографической камеры парами растворителей. Верхний слой используют для наполнения лодочки.
Отбор пробы
Воздух со скоростью 6 л/мин. протягивают через три пары параллельно соединенных поглотительных приборов Зайцева, содержащих по 2 мл поглотительного раствора. Отбирают не менее 60 л исследуемого воздуха при охлаждении. По мере улетучивания спирта в процессе отбора в поглотители периодически вносят спирт, подкисленный уксусной кислотой, до первоначального объема.
Ход анализа
Жидкость из первых трех поглотительных приборов переносят в выпаривательные чашки, смывают поглотительные приборы по 1 мл подкисленным спиртом, промывные жидкости объединяют с пробой. Аналогично поступают со вторыми поглотительными приборами.
Удаляют спирт выпариванием при комнатной температуре или при нагревании не выше 50 °C, так, чтобы осадок оставался влажным. Остаток в чашке количественно переносят в пробирку, обмывают чашку спиртом, подкисленным уксусной кислотой, и доводят объем до 0,3 мл.
В хроматографической камере по стенкам располагают фильтровальную бумагу, смоченную нижним слоем жидкости, полученной при приготовлении системы растворителей.
Верхний слой жидкости помещают в лодочку, которая расположена в хроматографической камере. Насыщение хроматографической камеры проходит в течение 3 — 4 ч. На лист хроматографической бумаги на расстоянии 7 см от нижнего края намечают линию старта, на которой фиксируют точки нанесения пробы, на расстоянии 3 — 3,5 см друг от друга.
С помощью микропипетки в указанные точки наносят пробу. В одну точку наносят содержимое первых поглотительных приборов, а во вторую — содержимое вторых, контрольных, поглотителей.
Наносят по 0,15 или 0,3 мл пробы в зависимости от содержания стирола. Если наносят всю пробу, то чашку обмывают этиловым спиртом. При нанесении растворов на бумагу ее обдувают теплым воздухом при помощи вентилятора.
Хроматографическую бумагу с пробой помещают в лодочку. Камеру герметизируют и оставляют при комнатной температуре на 14 — 15 ч. По истечении времени растворитель опускается на расстояние 25 см. Бумагу извлекают из камеры, высушивают на воздухе (0,5 — 1 ч) и орошают раствором дифенилкарбазида. Хроматограмму помещают в термостат и выдерживают в течение 1 — 2 мин. при 70 °C. При наличии стирола на хроматограмме проявляются окрашенные в фиолетовый цвет зоны локализации в виде круглых пятен (Rf = 0,8).
На линии старта также проявляются окрашенные зоны, свидетельствующие об избытке ацетата ртути. Зоны, характерные для стирола, вырезают из бумаги, измельчают и помещают в пробирки, в которые вносят по 3,5 мл очищенного этанола. Содержимое пробирок встряхивают, через 5 и 15 мин. элюаты фотометрируют на спектрофотометре в кюветах с толщиной слоя 1 см при длине волны 570 нм. Содержание стирола устанавливают по калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 45).
Таблица 45
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Стандартный раствор N 1, мл |
0 |
0,33 |
1,0 |
1,66 |
3,33 |
5 |
|
Поглотительный раствор, мл |
5 |
4,67 |
4,0 |
3,34 |
1,67 |
0 |
|
Содержание стирола в объеме 0,15 мл, мкг |
0 |
1,0 |
3,0 |
5,0 |
10,0 |
15,0 |
Согласно шкале стандартов (см. табл. 45), получают ряд растворов 1 — 2 — 3…5 с содержанием от 1 до 15 мкг стирола в объеме 0,15 мл.
На хроматографическую бумагу по линии старта наносят по 0,15 мл каждого раствора, подвергают хроматографированию и проводят все операции аналогично пробе. На основании данных оптических плотностей строят калибровочный график.
Шкалой стандартов можно пользоваться при визуальном определении.
Расчет см. выше.
![]() -МЕТИЛСТИРОЛ
-МЕТИЛСТИРОЛ
|
|
Мол. вес 118,18 |
Бесцветная или слегка желтоватая жидкость с неприятным запахом. Температура кипения 162 — 163 °C, удельный вес 0,914, в воде не растворим, растворим в органических растворителях.
Пары альфа-метилстирола вызывают раздражение глаз и верхних дыхательных путей. Жидкий ![]() -метилстирол вызывает раздражение кожных покровов.
-метилстирол вызывает раздражение кожных покровов.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ ![]() -МЕТИЛСТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ
-МЕТИЛСТИРОЛА ХРОМАТОГРАФИЕЙ НА БУМАГЕ
![]() -Метилстирол определяют аналогичным стиролу методом (см. выше).
-Метилстирол определяют аналогичным стиролу методом (см. выше).
ГИДРОПЕРЕКИСЬ ИЗОПРОПИЛБЕНЗОЛА
|
C6H5(CH3)2COOH |
Мол. вес 152,19 |
Жидкость бесцветная маслянистая, запах ее напоминает озон. Плотность 1,062 (20°), температура кипения 60 °C (0,2 мм), температура разложения 741°, воспламенения 80°. Взрывает при 170 °C. Растворима в органических растворителях, легко растворима в воде. Под действием кислот и катализаторов разлагается с образованием эквимолекулярных количеств фенола и ацетона.
Гидроперекись изопропилбензола вызывает раздражение слизистых оболочек, покраснение кожи, одышку, изменение состава крови.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,007 мг/м3.
ОПРЕДЕЛЕНИЕ ГИДРОПЕРЕКИСИ ИЗОПРОПИЛБЕНЗОЛА ПО ФЕНОЛУ [9]
Принцип определения
При действии серной кислоты на гидроперекись изопропилбензола образуется фенол, который взаимодействует с П-нитрофенилдиазонием и дает продукт, окрашенный в красный цвет. Чувствительность определения 1,5 мкг в определяемом объеме раствора. Мешают определению фенол, крезолы, амины.
Реактивы
1. Силикагель, КСМ, МСМ, КСК, МСК, размер зерен 0,25 — 0,5 мм.
2. Этанол, предварительно обработанный (см. выше).
3. Стандартный раствор гидроперекиси изопропилбензола (ГПИПБ). Исходный стандартный раствор готовят из 99,9% ГПИПБ в мерной колбе емкостью 25 мл.
В колбу вносят 5 мл этанола, взвешивают, затем добавляют 1 — 3 капли ГПИПБ и вновь взвешивают. Объем колбы доводят до метки этанолом. Расчетным путем устанавливают содержание его в 1 мл. Исходный раствор устойчив в течение месяца. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 100 мл. Для этого в колбу вносят расчетное количество исходного стандартного раствора 0,5 мл этанола и 2 мл серной кислоты уд. веса 1,84. Колбу нагревают на водяной бане при температуре 70° в течение часа, при этом колба должна находиться в наклонном положении и прикрыта обратной стороной притертой пробки. По отстаивании объем колбы доводят до метки водой. Раствор употребляют свежеприготовленный.
4. Соляная кислота, уд. вес 1,19.
5. Серная кислота, уд. вес 1,84.
6. Карбонат натрия насыщенный, 0,8% раствор.
7. Нитрит натрия, 25% раствор.
8. Паранитроанилин.
9. Паранитрофенилдиазоний. Готовят добавлением к 50 мл воды 2,5 мл соляной кислоты уд. веса 1,19, 2,5 мл 25% раствора нитрита натрия и 0,012 паранитроанилина. Тщательно перемешивают. Употреблять свежеприготовленный.
Отбор пробы
а) Для определения разовой концентрации отбор проб проводят в кипящий слой силикагеля.
Для этого по 3 мл силикагеля через воронку вносят в поглотительные приборы Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана. Два последовательно присоединенных прибора в свою очередь соединяют с поглотителем, служащим для улавливания переброшенных зерен силикагеля. Воздух протягивают со скоростью до 10 л/мин. в течение 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают со скоростью до 5 л/мин. через 2 последовательно соединенных поглотительных прибора, содержащих по 3 мл силикагеля. Отбор проб проводят непрерывно в течение суток.
Ход анализа
Силикагель из каждого поглотительного прибора отдельно переносят в пробирки с притертой пробкой.
Переброшенные зерна силикагеля присоединяют к содержимому второго поглотителя.
Добавляют по 4 мл этанола. Периодически в течение 30 мин. встряхивают содержимое пробирок. Для анализа берут по 2 мл прозрачного раствора и помещают их в пробирки с притертыми пробками, добавляют по 0,1 мл серной кислоты уд. веса 1,84 и нагревают пробирки при 70 °C в течение часа на водяной бане, поместив пробирки в наклонном положении и прикрыв их обратной стороной притертой пробки.
После охлаждения проводят нейтрализацию, добавляя по каплям насыщенный раствор карбоната натрия до щелочной реакции по лакмусу. Объем пробы доводят до 5 мл 0,8% раствором карбоната натрия. Добавляют 0,1 мл раствора n-нитрофенилдиазония. Содержимое перемешивают и через 10 мин. сравнивают окраску проб со шкалой стандартов (табл. 46), которую готовят одновременно и аналогично пробам.
Таблица 46
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,15 |
0,3 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
|
Насыщенный раствор карбоната натрия, мл |
До нейтрализации (по лакмусу) |
|||||||
|
Раствор карбоната натрия, 0,8%, мл |
До объема 5 мл |
|||||||
|
Содержание ГПИПБ, мкг |
1,5 |
3,0 |
4,0 |
6,0 |
8,0 |
10,0 |
15,0 |
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение бензола в воздухе. Заводская лаборатория, 1942, 9.
2. Алексеева М.В. Определение толуола в воздухе. Заводская лаборатория, 1942, 9.
3. Определение ксилола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., 1963, в. 7.
4. Алексеева М.В. Определение изопропилбензола. В кн.: Определение атмосферных загрязнений. М., Медгиз, 1963.
5. Алексеева М.В. Определение нитробензола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., 1964, в. 8.
6. Гронсберг Е.Ш. Определение этилбензола. В сб.: Определение вредных веществ в воздухе производственных помещений. Горький, 1960.
7. Казнина Н.И. Определение стирола в воздухе с помощью бумажной хроматографии. Гиг. и сан., 1968, 5.
8. Марич Ю.А., Борский В.И. Метод определения динила в воздухе производственных помещений. Гиг. и сан., 1971, 1.
9. Хрусталева В.А. Определение гидроперекиси изопропилбензола в воздухе. В сб.: Ученые записки Московского научно-исследовательского института санитарии и гигиены имени Ф.Ф. Эрисмана, 1960, 5.
Глава IX
ХЛОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Могут встречаться в виде газов, жидкостей и твердых веществ. Большая их часть — жидкости бесцветные, мало растворимы в воде, хорошо растворимы в спирте, эфире.
Хлорорганические соединения — углеводороды, являются наркотиками, некоторые действуют на внутренние органы (печень, почки), а также на нервную систему. Предельно допустимые концентрации некоторых хлорированных соединений даны в табл. 47.
Таблица 47
ПРЕДЕЛЬНО ДОПУСТИМЫЕ КОНЦЕНТРАЦИИ
|
Наименование |
Максимальная разовая, мг/м3 |
Среднесуточная, мг/м3 |
|
Дихлорэтан |
3 |
1 |
|
Трихлорэтилен |
4 |
1 |
|
Хлоропрен |
0,1 |
0,1 |
|
Четыреххлористый углерод |
4 |
2 |
ОПРЕДЕЛЕНИЕ ХЛОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ МЕТОДОМ СЖИГАНИЯ
В ПРИБОРЕ НИИ ГИГИЕНЫ ИМ Ф.Ф. ЭРИСМАНА [8]
Принцип определения
Хлорированные углеводороды сжигают в колонке с платиновой проволокой, накаленной докрасна (600 — 700 °C). Продукты сжигания поглощают раствором щелочи, в которой определяют ион хлора.
Чувствительность определения по хлору 1 мкг в анализируемом объеме. Хлориды, бромиды и йодиды мешают определению.
Аппаратура
Прибор для сжигания хлорированных соединений смонтирован на деревянной доске (схема показана на рис. 24). Состоит из двух частей: очистительной и аналитической. Аналитическая часть в свою очередь состоит из газовой пипетки емкостью 500 мл (7), стеклянной колонки для сжигания (2), в которую вмонтирована платиновая спираль (9) длиной 70 мм, сечением 0,3 мм, микропоглотителя (3) и уравнительной склянки на 550 мл (8). Микропоглотитель представляет собой пробирку высотой 45 — 50 мм, диаметром 12 мм (6), в которую вставлена стеклянная трубка (4) длиной 70 мм, диаметром 8 мм со шлифом в верхней части. Трубка соединена с колонкой сожжения, нижняя часть трубки оттянута и имеет диаметр 3 мм. В эту трубку помещена спираль из стеклянной палочки в 20 витков (5), длина спирали 50 — 52 мм, диаметр 7,5 мм. Спираль должна плотно входить в трубку. Это важно для полноты поглощения продуктов сожжения пробы.

Рис. 24. Схема прибора для определения
хлорированных углеводородов
Газовая пипетка (7) соединяется с колонкой сожжения посредством трехходового крана, который расположен вверху над пипеткой. Посредством этого крана можно соединить колонку с очистительной системой. На обратной стороне доски помещена очистительная система (1), состоящая из двух поглотительных приборов, первый прибор наполнен 5% раствором щелочи для поглощения хлористого водорода. Второй прибор содержит 0,01 н. раствор мышьяковистой кислоты для задержания хлора; газовая пипетка емкостью 500 мл. Она соединена с очистительной системой и посредством трехходового крана с колонкой сожжения; трансформатор мощностью 40 Вт.
Примечание. 1. Новый прибор, а также прибор после продолжительного употребления и после длительного перерыва в работе требует пропаривания. Для этого к прибору присоединяют парообразователь на 20 — 30 мин. После пропарки прибор осушают воздухом, не содержащим хлористого водорода и хлора. Колонку с платиновой спиралью включают в сеть только после того, как она высохнет. 2. Прибор для сжигания хлорированных углеводородов ПСУ изготавливают по МРТУ 42-2060-62 завод «Химлаборпосуда» (рис. 25).
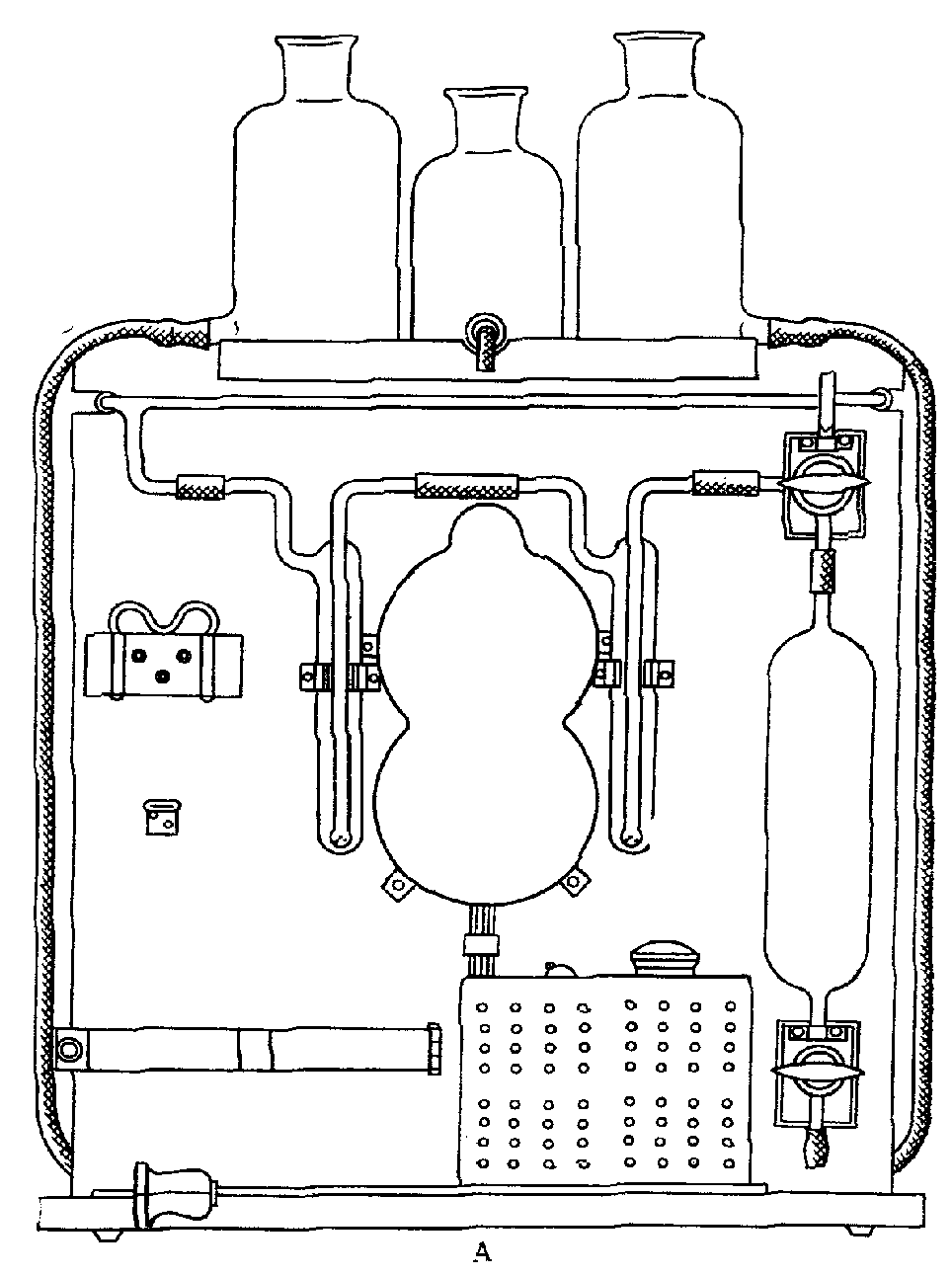
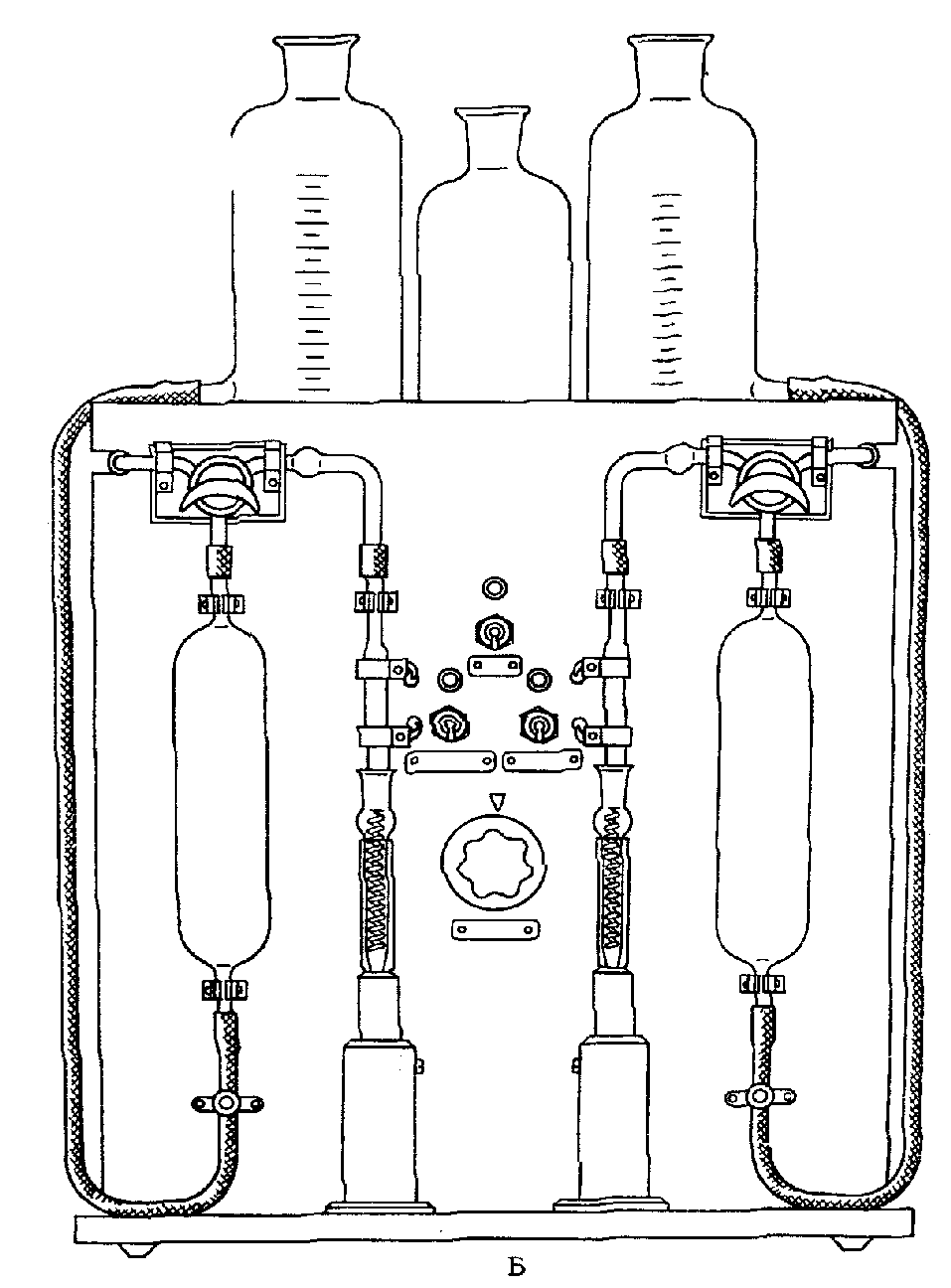
Рис. 25. Прибор ПСУ, изготовленный по МРТУ 42-2060-62
А — обратная сторона прибора; Б — общий вид
Реактивы
Все реактивы готовят на дважды перегнанной воде, свободной от иона хлора.
1. Поглотительный раствор — 0,01 н. раствор едкого натра.
2. Стандартный раствор: исходный раствор с содержанием 100 мкг/мл иона хлора, получают растворением навески хлорида калия. 0,0204 г соли растворяют в мерной колбе емкостью 100 мл. Рабочий стандартный раствор с содержанием 10 мкг/мл получают разбавлением исходного раствора в 10 раз.
3. Азотная кислота, уд. вес 1,34 — 1,4.
4. Нитрат серебра, 0,1% раствор: навеску 0,1 г нитрата серебра растворяют в 90 мл воды и в раствор вливают 10 мл азотной кислоты уд. веса 1,34 — 1,4.
5. Едкий натр, 5% и 10% растворы.
6. Сульфат натрия, насыщенный раствор.
7. Мышьяковистая кислота, 0,01 н. раствор 0,124 г мышьяковистого ангидрида растворяют в 1 мл 10% едкого натра, добавляют 200 мл воды и нейтрализуют осторожно 5% раствором серной кислоты в присутствии фенолфталеина. Раствор вливают в мерную колбу емкостью 250 мл. Объем раствора доводят до метки 5% раствором бикарбоната натрия.
8. Серная кислота, 5% раствор.
9. Бикарбонат натрия, 5% раствор.
Отбор пробы
Для определения разовой концентрации отбирают две пробы в газовые пипетки емкостью 500 — 1000 мл, предварительно наполненные насыщенным раствором сернокислого натрия. В месте отбора проб раствор выливают из пипеток в течение 5 — 10 мин. и газовая пипетка заполняется исследуемым воздухом. По окончании отбора проб зажимы на пипетках закрывают и в концы резиновых трубок вставляют стеклянные палочки.
Для определения среднесуточной концентрации отбирают пробу в течение суток при помощи аспиратора емкостью 3 л, заполненного насыщенным раствором сернокислого натрия. Скорость отбора 0,1 л/ч.
Подготовка прибора к анализу
Подготовка прибора к анализу состоит из следующих процессов: 1) промывка микропоглотителя, 2) проверка герметичности прибора, 3) продувание прибора воздухом, свободным от хлористого водорода и хлора, 4) контрольный анализ, т.е. проверка на чистоту прибора.
1. Микропоглотитель промывают хромовой смесью, затем водой и пропаривают его в течение 10 — 15 мин. После этого проверяют чистоту микропоглотителя на отсутствие иона хлора нитратом серебра в присутствии кислоты (азотной или серной). Если промывная вода помутнеет от нитрата серебра, микропоглотитель следует промывать и кипятить в воде и вновь проверить реакцию на ион хлора. Чистый микропоглотитель присоединяется к колонке для сожжения.
2. Проверка герметичности прибора заключается в том, что уравнительные склянки заполняют дистиллированной водой, помещают на верхнюю полку прибора, все краны и зажимы открывают, но предварительно на нижний конец микропоглотителя надевают резиновую трубку с плотно завернутым зажимом и с вставленной стеклянной палочкой на конце трубки.
Прибор можно считать герметичным, если вода из уравнительной склянки не поступает в пипетку.
3. Для продувания прибора воздухом, свободным от хлора и хлористого водорода, необходимо: А) заполнить газовую пипетку, помещенную на обратной стороне прибора, водой, а затем чистым воздухом продуть прибор; Б) заполнить газовые пипетки, помещенные на лицевой стороне прибора, водой, а затем очищенным воздухом.
Продуть прибор чистым воздухом.
А. При заполнении пипетки, помещенной на обратной стороне прибора, соединяют нижний конец ее с уравнительной склянкой, а верхний конец пипетки соединяют с помощью трехходового крана с колонкой сожжения. Уравнительную склянку помещают на верхнюю полку прибора. Осторожно открывают кран у нижнего конца пипетки, и вода поступает в пипетку, а воздух из пипеток выходит через колонку сожжения в помещение лаборатории.
При наполнении пипетки водой зажим у нижнего края закрывают и для заполнения ее воздухом уравнительную склянку помещают на стол, трехходовым краном пипетку соединяют с очистительной системой. Нижний зажим на пипетке открывают, вода вытекает из пипетки в уравнительную склянку, а воздух через очистительную систему наполняет пипетку.
Б. Заполняют водой пипетку, помещенную на лицевой стороне прибора. Уравнительные склянки соединяют с нижними концами пипеток, зажимы должны быть закрыты, помещают их на верхнюю полку. Трехходовыми кранами пипетки соединяют с колонками сожжения. Осторожно открывают зажимы у нижних концов пипеток, вода поступает в пипетки, а воздух через колонки выходит в помещение лаборатории. После наполнения пипеток водой нижние зажимы закрывают, уравнительные склянки помещают на стол, трехходовые краны повертывают и соединяют пипетки с очистительной системой. Зажимы у нижних концов пипетки открывают, вода постепенно вытекает в уравнительные склянки и воздух через очистительную систему наполняет пипетки. По окончании наполнения трехходовые краны повертывают на нейтральное положение.
В. После этого требуется продуть прибор чистым воздухом. Для этого уравнительные склянки помещают на верхние полки прибора. Пипетки, помещенные на лицевой стороне прибора, соединяют с колонками сожжения трехходовыми кранами. Нижние зажимы у пипеток осторожно открывают и вода поступает в пипетки, а воздух, проходя колонки сожжения и микропоглотитель, выходит в помещение лаборатории. Необходимо промыть прибор чистым воздухом с помощью пипетки, помещенной на обратной стороне прибора. После 2 — 3-кратного промывания проводят контрольный опыт.
4. При контрольном анализе, прибор заполнен чистым воздухом, газовую пипетку заполняют чистым воздухом, как описано выше. Микропоглотители снимают с прибора и заполняют 0,01 н раствором едкого натра, вливая 3 — 4 капли его на внутреннюю спираль микропоглотителя и 2 — 3 капли — в пробирку поглотителя (нижний конец внутренней трубки должен быть погружен в раствор). Микропоглотитель соединяют с колонками сожжения. Газовые пипетки на лицевой стороне прибора соединяют внизу с уравнительными склянками, заполненными насыщенным раствором сульфата натрия. Верхние концы пипеток соединяют с трехходовыми кранами встык. Кранами пипетки соединяют с колонками сожжения. Колонки включают в сеть, при этом платиновая спираль быстро нагревается до красного каления.
Уравнительные склянки помещают на верхнюю полку прибора и открывают сначала верхние зажимы газовых пипеток, затем нижние.
Раствор из уравнительных склянок поступает в пипетки, а воздух проходит через колонки сожжения и микропоглотители. Прохождение воздуха наблюдается в микропоглотителе в виде цепочки пузырьков воздуха. Скорость потока воздуха 20 — 25 мл/мин. После вытеснения воздуха из пипеток трехходовой кран повертывают на нейтральное положение, закрывают зажимы на пипетках, уравнительные склянки ставят на стол и соединяют с нижним концом газовой пипетки, помещенной на обратной стороне прибора.
Уравнительную склянку помещают на верхнюю полку и трехходовым краном соединяют пипетку с колонкой сожжения и осторожно открывают нижний зажим у пипетки. Раствор поступает в пипетку; воздухом промывают прибор в течение 5 — 10 мин.
По окончании промывки прибора выключают колонки из сети, отъединяют микропоглотители от колонок, промывают их поглотительным раствором. В перпендикулярно поставленный микропоглотитель вносят каждый раз по 5 — 6 капель раствора. Раствор из поглотителя переливают в колориметрические пробирки с меткой 1 мл. Операцию промывания повторяют несколько раз и промывной раствор сливают в 2 — 3 пробирки. Количество раствора в каждой пробирке должно быть 1 мл.
В пробирки вносят по 0,2 мл 0,1% раствора нитрата серебра в азотной кислоте. При отрицательной реакции на хлор прибор можно считать чистым и пригодным для проведения анализа. В противном случае контрольный анализ повторяют несколько раз.
Ход анализа
В микропоглотитель вносят 3 — 4 капли 0,01 н. раствора едкого натра через верхнюю часть и 2 — 3 капли этого раствора помещают в пробирку микропоглотителя, последний соединяют с колонкой сожжения. Газовую пипетку с пробой присоединяют к трехходовому крану встык, она должна быть в вертикальном положении. Нижний конец пипетки соединяют с уравнительной склянкой. Трехходовой кран подвертывают так, чтобы пипетка с пробой была соединена с колонкой сожжения. Колонку включают в сеть и нагревают до красного каления и после этого открывают верхний зажим пипетки с пробой, затем открывают нижний зажим у пипетки, воздух из пипетки пропускают со скоростью 20 — 25 мл/мин. Далее поступают так, как указано в контрольном анализе.
После промывания микропоглотителя в колориметрические пробирки наливают по 0,2 мл 0,1% раствора нитрата серебра и помутнение проб сравнивают через 5 мин. после вливания нитрата со шкалой стандартов, которую готовят одновременно с пробой (табл. 48).
Таблица 48
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание хлора, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет концентраций хлорорганических соединений (мг/м3) воздуха проводят по формуле:
![]()
где: a — количество хлора в пробе (мкг); к — коэффициент пересчета хлора на искомое соединение; V0 — объем воздуха в литрах, отобранного для анализа, приведенного к нормальным условиям.
Примечания. 1. Для нижеуказанных веществ даны коэффициенты пересчета с хлора на: дихлорэтан — 1,4, четыреххлористый углерод — 1,08, хлороформ — 1,13, трихларэтилен — 1,23, тетрахлорэтан — 1,01, хлоропрен — 2,49.
2. В случае необходимости определения других хлорорганических веществ коэффициент пересчета устанавливают в каждом случае отдельно, сжигая известные количества вещества, внесенные в специальный «гусек».
МЕТА- И ПАРАХЛОРАНИЛИНЫ
Метахлоранилин — жидкость, не растворимая в воде, температура кипения 230 °C, плотность при 20 °C 1,216, хорошо растворима в этаноле и эфире. Оба изомера обладают запахом, напоминающим нафталин.
Хлоранилины — вещества ядовитые, токсичные, их действие сходно с анилином.
Предельно допустимые концентрации парахлоранилина: максимальная разовая 0,04 мг/м3, среднесуточная 0,01 мг/м3.
Предельно допустимая концентрация метахлоранилина среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ПАРА- и МЕТАХЛОРАНИЛИНОВ
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [3]
Принцип определения
При взаимодействии м- и п-хлоранилинов с парадиметиламинобензальдегидом в кислой среде образуется соединение, окрашенное в желтый цвет.
Чувствительность определения 0,25 мкг в анализируемом объеме раствора. Непредельные углеводороды, высшие спирты, амины мешают определению.
Реактивы
1. Поглотительный раствор — 10% раствор уксусной кислоты.
2. Стандартный раствор хлоранилина. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 15 — 20 мл поглотительного раствора. Колбу взвешивают и в нее вносят несколько кристаллов или капель хлоранилина свежеперекристаллизованного или перегнанного и вновь взвешивают. По разности двух взвешиваний устанавливают количество хлоранилина в колбе и рассчитывают его содержание в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вливают такое количество исходного раствора, которое содержит 500 мкг хлоранилина, и объем жидкости доводят до метки поглотительным раствором.
3. Парадиметиламинобензальдегид, 2% раствор в 40% растворе уксусной кислоты.
4. Уксусная кислота, 40% раствор.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через два соединенных поглотительных прибора с пористыми фильтрами N 1, наполненных по 3 мл поглотительного раствора в каждом. Продолжительность отбора 20 — 30 мин. при охлаждении.
б) Для определения среднесуточной концентрации воздух протягивают со скоростью 0,3 л/мин. через поглотительные приборы непрерывно в течение суток. Допустимо отбор проб проводить 6 — 12 раз с перерывами 4 — 2 ч в одни и те же поглотительные приборы.
Ход анализа
Анализируют пробу из каждого поглотительного прибора отдельно.
2 мл пробы переносят в пробирки, вливают по 0,6 мл 25% раствора парадиметиламинобензальдегида, содержимое пробирок взбалтывают и через 5 мин. измеряют оптическую плотность при длине волны 445 нм в кювете с толщиной слоя 0,5 см по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание хлоранилина определяют по предварительно построенному калибровочному графику. Готовят шкалу стандартов (табл. 49).
Таблица 49
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,025 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
2 |
1,975 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание хлоранилина, мкг |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ХЛОРБЕНЗОЛ
Бесцветная жидкость с температурой кипения 132°, плотностью при 20 °C 1,106, мало растворимая в воде, но хорошо растворимая в стироле и других органических растворителях.
Хлорбензол — наркотик, действует на кроветворные органы, но слабее, чем бензол.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,10 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРБЕНЗОЛА НИТРОВАНИЕМ [1]
Принцип определения
При нитровании хлорбензола образуется нитросоединение, которое в щелочной среде в присутствии кетона дает соединение, окрашенное в розовый цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора.
Ароматические соединения мешают определению.
Реактивы
1. Стандартный раствор хлорбензола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 2 — 3 мл нитрационной смеси, и колбу взвешивают. Затем в колбу вносят 2 — 3 капли хлорбензола, вновь взвешивают, раствор встряхивают и помещают колбу в кипящую водяную баню на 40 мин. После охлаждения раствор доводят до метки нитрационной смесью. По разности двух взвешиваний устанавливают количество хлорбензола в 1 мл раствора.
Рабочий стандартный раствор с содержанием 100 мкг/мл готовят соответствующим разведением исходного раствора.
Другие реактивы и отбор пробы см. «Определение бензола нитрованием» выше.
Ход анализа
Поглотительный прибор с пробой помещают в кипящую водяную баню на 40 мин. По охлаждении раствор переводят в коническую колбу емкостью 50 мл и поглотительный прибор ополаскивают несколько раз 5 — 6 мл воды. Промывную воду присоединяют к пробе, избегая сильного разогревания раствора. После охлаждения раствор нейтрализуют 25% раствором аммиака. Нейтрализацию проводят, пользуясь бюреткой с краном в присутствии лакмусовой бумаги. Затем пробу переносят в делительную воронку и добавляют 2,2 мл бутанона, смесь в воронке встряхивают в течение 5 мин. до разделения слоев. Бутаноновый раствор переливают и объем доводят бутаноном до 2 мл, прибавляют 0,75 мл щелочи. Содержимое пробирки встряхивают в течение нескольких минут и через 20 — 30 мин. сравнивают интенсивность пробы со шкалой стандартов. Одновременно готовят раствор для стандартной шкалы. Для этого берут 1 мл рабочего стандартного раствора, выливают в коническую колбу емкостью 100 мл, в которую внесено 8 — 10 мл воды. Раствор охлаждают и нейтрализуют 25% раствором аммиака в присутствии лакмуса.
Охлажденный нейтральный раствор выливают в делительную воронку и добавляют 10 мл бутанона. Раствор встряхивают в течение 10 мин. и оставляют в покое до полного разделения слоев. Нижний слой, водный, сливают через кран, верхний, бутаноновый, переливают через верх воронки в мерную колбу емкостью 10 мл или в пробирку с притертой пробкой и меткой 10 мл. Объем раствора доводят до метки бутаноном. Получают стандартный раствор в бутаноне с содержанием 10 мкг/мл. Из этого раствора готовят шкалу стандартов (табл. 50).
Таблица 50
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный бутаноновый раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бутанон, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание хлорбензола, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
НИТРОХЛОРБЕНЗОЛЫ
Нитрохлорбензолы орто-, мета-, пара-; представляют собой кристаллические вещества с запахом нитробензола.
Относятся к токсическим соединениям.
Оказывают действие на нервную систему, печень и др. Действие их сходно с действием нитробензола.
Предельно допустимые концентрации пара- и ортонитробензолов; среднесуточная 0,004 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТА-, ПАРАНИТРОХЛОРБЕНЗОЛА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [4]
Принцип определения
Метод основан на восстановлении нитрохлорбензола до хлоранилина, который при взаимодействии с п-диметиламинобензальдегидом образует соединение, окрашенное в желтый цвет.
Чувствительность определения — 0,2 мкг в анализируемом объеме раствора.
Анилин, хлоранилин, дихлоранилин и другие ароматические амины мешают определению, а также спирты и альдегиды.
Реактивы, отбор пробы, ход анализа см. «Определение хлоранилина с парадиметиламинобензальдегидом» выше.
МЕТА- И ПАРАХЛОРФЕНИЛИЗОЦИАНАТЫ
|
ClC6H4(NCO) |
Мол. вес 153,58 |
Метахлорфенилизоцианат — бесцветная жидкость, не растворима в воде, слабо растворима в водных растворах щелочи.
Парахлорфенилизоцианат — твердое кристаллическое вещество белого цвета, очень гигроскопично, обладает резким запахом, в воде не растворимо и трудно растворимо в водных растворах щелочи.
Хлорфенилизоцианаты токсичны.
Предельно допустимые концентрации: максимальная разовая и среднесуточная для парахлорфенилизоцианата 0,0015 мг/м3, для метахлорфенилизоцианата 0,005 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТА- и ПАРАХЛОРФЕНИЛИЗОЦИАНАТОВ
ПО ОБРАЗОВАНИЮ ХЛОРАНИЛИНА [5]
Принцип определения
При взаимодействии со щелочью мета- и парахлорфенилизоцианат гидролизуется с образованием хлоранилина, который диазотируют и сочетают с ![]() -нафталом, при этом образуется продукт, окрашенный в розовый цвет.
-нафталом, при этом образуется продукт, окрашенный в розовый цвет.
Чувствительность определения — 0,1 мкг в анализируемом объеме раствора.
Анилин, мета- и парахлоранилин, фенилизоцианат мешают определению.
Реактивы
1. Поглотительный раствор — 0,5 н. раствор едкого натра.
2. Стандартный раствор мета- или парахлорфенилизоцианата. Готовят исходный раствор, для этого в мерную колбу емкостью 25 — 50 мл наливают 15 — 20 мл 0,5 н. раствора едкого натра, взвешивают, вносят в нее несколько кристалликов продукта, снова взвешивают. По разности двух взвешиваний устанавливают количество хлорфенилизоцианата в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят путем соответствующего разведения исходного раствора.
Раствор применяют свежеприготовленный.
3. Аммиак, 0,5% раствор.
4. Уксусная кислота, 1 н. раствор.
5. Нитрит натрия, 7% раствор.
6. Бромид натрия, 12% раствор.
7. Составной раствор: растворы нитрита натрия и бромида натрия смешивают в отношении 1:1.
8. Едкий натр, 20% раствор.
9. Этанол 96°.
10. ![]() -Нафтол, 0,05% раствор в этаноле.
-Нафтол, 0,05% раствор в этаноле.
Отбор пробы
Воздух со скоростью 1 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористым фильтром N 1, наполненных по 1,5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. Анализ проб проводят в день отбора.
Ход анализа
Содержимое каждого поглотительного прибора анализируют отдельно. 1 мл пробы помещают в пробирку с притертой пробкой с меткой 5 — 10 мл. В пробирку с пробой наливают 1 мл 0,5% раствора аммиака, 0,9 мл 1 н. раствора уксусной кислоты, объем раствора в пробирке доводят до 5 мл водой, раствор встряхивают и добавляют 0,3 мл составного раствора. Через 5 мин. в пробирку наливают 0,1 мл 20% раствора едкого натра, раствор перемешивают и в него вливают 0,5 мл 0,05% раствора альфа-нафтола и 1 мл этанола. Содержимое пробирки встряхивают и оставляют в покое на 50 — 60 мин.
По истечении этого времени измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 508 нм.
Содержание м-п-хлорфенилизоцианата в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 51).
Таблица 51
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,01 |
0,03 |
0,05 |
0,07 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,99 |
0,97 |
0,95 |
0,93 |
0,9 |
0,7 |
0,5 |
0,8 |
0 |
|
Содержание МХФЦ или ПХФЦ, мкг |
0 |
0,1 |
0,3 |
0,5 |
0,7 |
1,0 |
3,0 |
5,0 |
7,0 |
10 |
Все пробирки обрабатывают аналогично пробам. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ЭПИХЛОРГИДРИН
|
|
Мол. вес 117 |
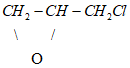
Прозрачная бесцветная жидкость, температура кипения 117 °C, плотность 1,18, мало растворима в воде, но хорошо растворима в этаноле и эфире.
Эпихлоргидрин действует раздражающе на слизистые оболочки дыхательных путей.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ ЭПИХЛОРГИДРИНА ПО ФОРМАЛЬДЕГИДУ [7, 10]
Принцип определения
При взаимодействии эпихлоргидрина с йодной кислотой в сернокислой среде образуется формальдегид, который определяют с хромотроповой кислотой.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Формальдегид, окись этилена, этиленгликоль мешают определению.
Реактивы
1. Поглотительный раствор — серная кислота, 60% раствор (по весу).
2. Стандартный раствор эпихлоргидрина. Готовят исходный стандартный раствор: в мерную колбу емкостью 50 мл вливают 10 — 15 мл поглотительного раствора и взвешивают. Затем в колбу вносят 2 капли эпихлоргидрина и вновь взвешивают. По разности двух взвешиваний устанавливают количество эпихлоргидрина в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл эпихлоргидрина получают соответствующим разбавлением исходного.
3. Перйодат калия, 1% раствор в 60% серной кислоте.
4. Сульфит натрия, 40% раствор готовят при нагревании. Устойчив в течение 2 — 3 дней.
5. Хроматроповая кислота, 0,3 г кислоты растворяют в 5 мл воды, затем осторожно приливают 95 мл серной кислоты удельного веса 1,84. Раствор устойчив в течение недели при хранении в темном прохладном месте.
6. Силикагель ACM.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, наполненный 6 мл поглотительного раствора.
При отборе проб на твердый сорбент по 2 мл силикагеля помещают в два поглотительных прибора Института имени Ф.Ф. Эрисмана. Отбор проводят в «кипящий» слой. Скорость отбора 5 л/мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,1 — 0,2 л/мин. протягивают через поглотительный прибор непрерывно в течение суток. В первом и втором случае отбор проводят в течение 20 — 30 мин.
Допустимо отбор проводить 6 — 12 раз с перерывами 4 — 2 ч по 20 — 30 мин. в один и тот же поглотительных прибор.
Отбор среднесуточных проб можно проводить в кипящий слой силикагеля, скорость до 5 л/мин.
Ход анализа
В пробирку помещают 5 мл пробы, в нее вносят 1 каплю перйодата калия, раствор перемешивают и пробирку помещают на 15 мин. в кипящую водяную баню. После охлаждения раствора прибавляют осторожно 5 капель сульфита и тщательно перемешивают. Раствор должен обесцветиться. Избытка сульфита необходимо избегать. Через 2 — 3 мин. в пробирку приливают 1 мл хромотроповой кислоты. Раствор перемешивают и помещают на 30 мин. в кипящую водяную баню. После охлаждения раствора в пробирку вливают 2 мл воды и через 20 — 30 мин. измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 570 нм.
Содержание эпихлоргидрина в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 52).
Таблица 52
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,2 |
1,6 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
3,8 |
3,4 |
3,0 |
|
Содержание эпихлоргидрина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
12 |
16 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят одновременно с пробой.
Ход анализа после отбора проб на силикагель. В каждую пробирку с пробой наливают по 4 мл поглотительного раствора и содержимое пробирок встряхивают. Затем готовят шкалу стандартов, как указано в табл. 52, но предварительно в каждую пробирку шкалы всыпают по 2 мл силикагеля. Вносят стандартный раствор и содержимое пробирок встряхивают. После этого объем в каждой пробирке доводят до 4 мл поглотительным раствором. Далее пробу и шкалу обрабатывают так, как при отборе пробы в жидкую поглотительную среду.
Расчет см. выше.
ЧЕТЫРЕХХЛОРИСТЫЙ УГЛЕРОД
Бесцветная, легко подвижная, летучая жидкость с неприятным запахом. Температура кипения 76,8°, плотность при 2° 1,595, мало растворима в воде, легко растворима в этаноле, эфире, ацетоне.
Четыреххлористый углерод является наркотиком, вызывает поражение печени, почек. Предельно допустимая максимальная разовая концентрация 4 мг/м3, среднесуточная 2 мг/м3.
ОПРЕДЕЛЕНИЕ ЧЕТЫРЕХХЛОРИСТОГО УГЛЕРОДА С ПИРИДИНОМ [6]
Принцип определения
При взаимодействии четыреххлористого углерода с пиридином образуется соединение, которое с анилином в присутствии ацетона дает дианилид глютаконового альдегида.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Хлороформ, хлораль и некоторые другие галоидоуглеводороды мешают определению. Хлористый метил, хлористый метилен, дихлорэтан, спирты, хлористый водород не мешают определению.
Реактивы
1. Поглотительный раствор — пиридин. Пиридин применяют свежеперегнанный, для этого его кипятят в течение часа с обратным холодильником над кристаллической щелочью (8 — 10 г на 100 мл).
Перегоняют, добавив кристаллической щелочи, и отбирают фракцию, кипящую при 114 — 116 °C. Хранят в стеклянной посуде в темном месте.
2. Стандартный раствор четыреххлористого углерода. Исходный стандартный раствор готовят растворением точной навески четыреххлористого углерода в пиридине.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разведением исходного раствора. Рабочий стандартный раствор устойчив в течение 6 ч.
3. Анилин, х.ч., свежеперегнанный.
4. Ацетон, х.ч.
5. Уксусная кислота, ледяная.
6. Натр едкий, 0,5 н. раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через два поглотительных прибора с пористым фильтром N 1, наполненные по 3 мл пиридина.
Продолжительность отбора 7 — 10 мин.
При наполнении приборов отмечают уровень жидкости и периодически доводят его до первоначального пиридином. Во время отбора поглотительные приборы охлаждают водой со льдом.
Ход анализа
Содержимое приборов количественно переносят в колориметрическую пробирку, добавляют 0,1 мл ацетона, 0,2 мл 0,5 н. раствора щелочи, перемешивают и нагревают в течение 4 мин. при 100 °C.
По охлаждении вносят 0,5 мл уксусной кислоты, 0,1 мл анилина и разбавляют смесь водой до 4 мл.
Через 15 мин. окрашенный в оранжевый цвет раствор фотометрируют при длине волны 485 — 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание четыреххлористого углерода в анализируемом объеме определяют по предварительно калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 53).
Таблица 53
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
2 |
|
Поглотительный раствор, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
0 |
|
Содержание четыреххлористого углерода, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения. Окраска растворов устойчива в течение 3 ч.
Расчет см. выше.
ХЛОРОФОС
|
|
Мол. вес 257,5 |
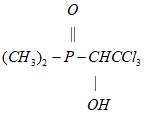
Твердое вещество с температурой плавления 81 °C, температурой кипения 120 °C (0,4 мм рт. ст.), легко растворимое в воде. Хлорофос используют как инсектофунгицид. Обладает токсическими свойствами.
Предельно допустимые концентрации: максимальная разовая 0,04 мг/м3, среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ХЛОРОФОСА ПО ХЛОРУ [9]
Принцип определения
Метод основан на омылении хлорофоса раствором щелочи при нагревании и определении иона хлора по реакции с нитратом серебра.
Чувствительность определения — 2 мкг хлорофоса в анализируемом объеме раствора. Определению мешают вещества, дающие реакцию с нитратом серебра.
Реактивы
1. Стандартный раствор с содержанием 100 мкг/мл хлорофоса. Готовят растворением 0,01 г хлорофоса в 100 мл бидистиллированной воды.
2. Едкий натр, 10% раствор.
3. Азотная кислота, 20% раствор.
4. Нитрат серебра, 1% раствор.
Все реактивы готовят на бидистиллированной воде.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 3 — 4 л/мин. протягивают через систему, состоящую из патрона с фильтром АФА-XII и двух поглотительных приборов Рыхтера (малая модель), содержащих по 4 мл бидистиллированной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
Фильтр переносят в стакан и промывают 2 раза водой, доводят объем до 4 мл. Пробу из поглотительных приборов переносят в пробирки с притертыми пробками. К пробам приливают по 1 мл 10% раствора едкого натра и нагревают на кипящей водяной бане в течение 30 мин. По охлаждении прибавляют по 1 мл 10% раствора азотной кислоты и 0,1 мл 5% раствора нитрата серебра, тщательно взбалтывают и через 10 мин. нефелометрируют в кюветах с толщиной слоя 1 см при длине волны 364 нм по отношению к контролю, который строят одновременно и аналогично пробам.
Содержание хлорофоса в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 54).
Таблица 54
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
|
Поглотительный раствор, мл |
2 |
1,98 |
1,95 |
1,9 |
1,8 |
1,7 |
1,6 |
1,5 |
|
Содержание хлорофоса, мкг |
0 |
2 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ХЛОРОПРЕН
(Хлористый дивинил, 2-хлор-1,3-бутадиен)
|
CH2=CH-CCl=CH2 |
Мол. вес 88,53 |
Бесцветная жидкость с температурой кипения 59,4 °C. Плотность 0,958 (20 °C), упругость пара 176,2 мм рт. ст. (20 °C). Хлоропрен легко полимеризуется, плохо растворим в воде, растворим в органических растворителях. Хлоропрен токсичен, раздражает дыхательные пути, вызывает дерматиты. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,10 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ХЛОРОПРЕНА [2]
Принцип определения
Метод основан на измерении оптической плотности спиртового раствора хлоропрена при длине волны 222,6 нм. Чувствительность определения — 0,5 мкг хлоропрена в анализируемом объеме раствора. Определению мешают вещества, поглощающие при длине волны 222,6 нм.
Реактивы
1. Поглотительный раствор — этиловый спирт 96°, ректификат.
2. Стандартные растворы хлоропрена. Исходный стандартный раствор хлоропрена готовят следующим образом. В мерную колбу емкостью 25 мл вносят 10 — 15 мл этилового спирта, взвешивают, затем вносят несколько капель свежеперегнанного хлоропрена и снова взвешивают. Объем в колбе доводят до метки спиртом, тщательно перемешивают и по разности между первым и вторым взвешиванием рассчитывают содержание хлоропрена в 1 мл раствора. Соответствующим разведением готовят стандартный раствор с содержанием 100 мкг/мл, а из него рабочий стандартный раствор с содержанием 10 мкг/мл хлоропрена. Растворы готовят в день анализа.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 5 л/ч протягивают через 3 — 4 поглотительных прибора с пористой пластинкой N 1, содержащих по 10 мл этилового спирта. Во время отбора поглотительные приборы помещают в сосуд со льдом.
Ход анализа
Содержимое каждого поглотительного прибора переливают в кварцевую кювету с толщиной слоя 1 см и измеряют оптическую плотность на спектрофотометре при длине волны 222,6 нм.
В качестве контроля используют этиловый спирт. Содержание хлоропрена в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 55).
Таблица 55
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,8 |
1,6 |
3,2 |
|
Этиловый спирт, мл |
10 |
9,95 |
9,9 |
9,8 |
9,6 |
9,2 |
8,4 |
6,8 |
|
Содержание хлоропрена, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
8,0 |
16,0 |
32,0 |
Растворы шкалы стандартов фотометрируют в тех же условиях, что и пробы.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение хлорбензола. В кн.: Определение вредных веществ в воздухе промышленных помещений. М., Госхимиздат, 1954.
2. Акопян И.Х., Абешян М.М. и др. Спектрофотометрический метод определения хлоропрена в атмосферном воздухе. Гиг. и сан., 1970, 9.
3. Андреещева Н.Г. Простые методы определения микроконцентраций анилина, метахлоранилина, парахлоранилина и 3,4-дихлоранилина в атмосферном воздухе. Гиг. и сан., 1968, 11.
4. Андреещева Н.Г. Определение микроконцентраций метанитрохлорбензола, п-нитрохлорбензола и нитробензола в атмосферном воздухе. Гиг. и сан., 1968, 12.
5. Беляков А.А., Тубина А.Я. Определение мета- и парахлорфенилизоционата. В сб.: Новое в области промышленно-санитарной химии. М., «Медицина», 1969.
6. Беляков А.А. Определение четыреххлористого углерода. В кн.: Определение вредных веществ в воздухе производственных помещений, г. Горький, 1970.
7. Гронсберг Е.Ш. Определение эпихлоргидрина. В сб.: Технические условия на методы определения вредных веществ в воздухе. М., 1964, в. 3.
8. Сендерихина Д.П. Определение хлорированных углеводородов в воздухе методом микросжигания. Гиг. и сан., 1954, 8.
9. Табакова С.А. Экспериментальные данные к гигиеническому нормированию хлорофоса. Гиг. и сан., 1969, 11.
10. Фомин А.П. Биологическое действие эпихлоргидрина и его гигиеническое значение как фактора загрязнения атмосферы. Гиг. и сан., 1966, 9.
Глава X
СПИРТЫ, ФЕНОЛЫ, ЭФИРЫ
СПИРТЫ ЖИРНОГО РЯДА
Спирты одноатомные насыщенные.
Физические свойства и предельно допустимые концентрации некоторых спиртов жирного ряда приведены в табл. 56.
Таблица 56
|
Наименование спирта |
Формула |
Молекулярный вес |
Состояние в обычных условиях |
Удельный вес (20°) |
Точка кипения °C |
ПДК, мг/м3 |
|
|
максимальная разовая |
среднесуточная |
||||||
|
Метиловый |
CH3OH |
32,04 |
Жидкость |
0,792 |
66 |
1,0 |
0,5 |
|
Этиловый |
C2H5OH |
46,03 |
То же |
0,789 |
78,4 |
5,0 |
5,0 |
|
Пропиловый |
C3H7OH |
60,09 |
« |
0,804 |
96 — 98 |
0,3 |
0,3 |
|
Изопропиловый |
C3H7OH |
60,09 |
« |
0,786 |
79,5 — 81,5 |
0,6 |
0,6 |
|
Бутиловый |
C4H9OH |
74,12 |
« |
0,810 |
118 |
0,1 |
0,1 |
|
Амиловый |
C5H11OH |
88,15 |
« |
0,802 |
137,7 |
— |
— |
|
Изооктиловый |
C8H17OH |
130,23 |
« |
0,827 |
194 |
0,15 |
0,15 |
Одноатомные насыщенные спирты обладают выраженными наркотическими свойствами. Сила наркотического действия возрастает с увеличением молекулярного веса. Пары спиртов обладают также раздражающим действием на слизистые оболочки.
Метиловый спирт занимает особое место среди спиртов этого ряда, так как в организме он дает весьма токсичные соединения. Это сильный нервный и сосудистый яд, обладающий кумулятивным действием. Для него весьма характерным является поражение зрительного нерва и сетчатки глаза.
МЕТИЛОВЫЙ СПИРТ (МЕТАНОЛ)
ОПРЕДЕЛЕНИЕ МЕТИЛОВОГО СПИРТА ПО ФОРМАЛЬДЕГИДУ [2]
Принцип определения
Метод основан на окислении метилового спирта до формальдегида, который определяют по реакции с хромотроповой кислотой.
Чувствительность фотометрического определения — 4 мкг метилового спирта в анализируемом объеме раствора, чувствительность визуального определения — 0,4 мкг.
Формальдегид мешает определению.
Реактивы
1. Поглотительный раствор: дважды перегнанная вода.
2. Стандартные растворы метилового спирта. Исходный раствор готовят так: в мерную колбу емкостью 50 мл вливают 10 мл дважды перегнанной воды и взвешивают на аналитических весах. Затем вносят в колбу 2 — 3 капли метанола и снова взвешивают, по разности весов определяют количество метанола. Раствор в колбе доводят до метки водой. Вычисляют содержание метанола в 1 мл раствора. Из этого раствора готовят раствор с содержанием 10 мг/мл. Перед определением готовят рабочий стандартный раствор с содержанием в 1 мл 10 мкг метанола.
3. Серная кислота 9 М. К 530 мл дистиллированной воды осторожно приливают 470 мл серной кислоты удельного веса 1,82.
4. Серная кислота 15 М. К 100 мл воды осторожно приливают 900 мл серной кислоты удельного веса 1,80 — 1,82.
5. Перманганат калия, 1% раствор.
6. Хромотроповая кислота (1,8-диоксинафталин, 3,6-дисульфокислота). 0,5 г хромотроповой кислоты растворяют в 10 мл воды, раствор применяют свежеприготовленным. Для определения готовят следующий раствор: берут 2 мл раствора хромотроповой кислоты в колбу емкостью 50 мл и доводят раствор до метки 15 М серной кислотой.
7. Сульфит натрия, 3% раствор, применяют свежеприготовленным.
8. Фосфорная кислота, 85% раствор, уд. вес 1,71.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 5 мл дважды перегнанной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора переносят в пробирку 2 мл пробы, добавляют по 0,25 мл фосфорной кислоты, а затем по 2 капли 1% раствора перманганата калия и помещают точно на 15 мин. в баню со льдом. По истечении этого времени пробирки вынимают из бани и в них по каплям вносят 3% раствор сульфита, до обесцвечивания раствора, избегая избытка сульфита. Затем во все пробирки вливают по 2 мл хромотроповой кислоты и помещают их на 30 мин. в кипящую водяную баню. По охлаждении в пробирки наливают 9 М серную кислоту до объема 5 мл, перемешивают и через 50 — 60 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 574 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание метанола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 57).
Таблица 57
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Вода, мл |
2 |
1,6 |
1,4 |
1,2 |
1,0 |
0 |
|
Содержание метанола, мкг |
0 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам и строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами (табл. 58).
Таблица 58
ШКАЛА СТАНДАРТОВ ДЛЯ ВИЗУАЛЬНОГО ОПРЕДЕЛЕНИЯ МЕТАНОЛА
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,04 |
0,06 |
0,08 |
0,1 |
0,2 |
|
Вода, мл |
2 |
1,96 |
1,94 |
1,92 |
1,9 |
1,8 |
|
Содержание метанола, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
Расчет см. выше.
———————————
<1> Раздельное определение метилового спирта и формальдегида см. ниже.
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА СПИРТОВ
ГРУППЫ C1 — C10 С ВАНАДИЙ-ОКСИХИНОЛИНОВЫМ КОМПЛЕКСОМ
(МЕТИЛОВОГО, ЭТИЛОВОГО, ПРОПИЛОВОГО, БУТИЛОВОГО,
АМИЛОВОГО, ГЕКСИЛОВОГО, ГЕПТИЛОВОГО, ОКТИЛОВОГО,
НОНИЛОВОГО, ДЕЦИЛОВОГО) [8]
Принцип определения
При взаимодействии спиртов с ванадий-оксихинолиновым комплексом образуется соединение, окрашенное в розово-оранжевый цвет. Избыток ванадий-оксихинолинового комплекса, окрашенный в черный цвет, разрушают щелочью.
Чувствительность — 5 мкг спирта в определяемом объеме раствора.
Не мешают определению ацетон, формальдегид, динил, кислоты жирного ряда (муравьиная, уксусная, масляная и др.), бутилацетат, метилацетат, винилацетат, сложные эфиры ортофосфорной кислоты (бутиловый, диметиловый, гептиловый, октиловый, нониловый) в количестве до 2 мг.
Реактивы
1. Стандартные растворы отдельных спиртов. Исходные стандартные растворы с содержанием 1 мг/мл готовят растворением расчетных количеств спиртов в бензоле. Рабочие стандартные растворы с содержанием 100 мкг спирта в 1 мл готовят разбавлением исходных растворов в 10 раз бензолом. Стандартные растворы сохраняются в течение суток. Для приготовления стандартного раствора используют спирт, который содержится в воздухе в большем количестве или наиболее токсичен.
2. Бензол х.ч. Очищают бензол следующим образом: к 900 мл бензола добавляют 100 мл серной кислоты уд. веса 1,84, помещают смесь в делительную воронку и встряхивают. Повторяют операцию до тех пор, пока кислота не перестанет окрашиваться или будет иметь слабо желтую окраску. После удаления кислоты бензол дважды встряхивают с водой, затем с 10% раствором едкого натра и снова с водой до нейтральной реакции (по фенолфталеину). Очищенный бензол осушают прокаленным хлористым кальцием в течение 1 — 2 сут., фильтруют и хранят в закрытой склянке.
3. Уксусная кислота, 5% раствор.
4. 8-оксихинолин, 2% раствор в 5% растворе уксусной кислоты.
5. Ванадат аммония, 0,08% раствор.
6. Едкий натр, 1 н. раствор.
Реактивы устойчивы в течение 3 мес.
7. Ванадий-оксихинолиновый комплекс. Готовят непосредственно перед использованием. В делительную воронку вносят 2,4 мл 5% раствора уксусной кислоты, 1,2 мл раствора 8-оксихинолина, 4 мл раствора ванадата аммония. Содержимое воронки перемешивают, затем добавляют 8 мл бензола и встряхивают смесь растворов в течение 0,5 мин. После разделения слоев нижний, водный, удаляют. Верхний слой, представляющий собой раствор ванадий-оксихинолинового комплекса в бензоле, сливают в сухую пробирку с пробкой.
8. Активированный уголь марки АГ-5 обрабатывают концентрированной соляной кислотой в течение 1 — 1 1/2 г ч при кипячении. Затем кислоту сливают, а уголь промывают несколько раз дистиллированной водой, 0,1 н. раствором аммиака до тех пор, пока последний перестанет окрашиваться. Далее уголь снова промывают дистиллированной водой до отрицательной реакции на ион хлора и сушат тонким слоем при 100 — 120 °C в течение 1 — 2 ч. Хранят уголь в склянке с притертой пробкой.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительную трубку (см. рис. 6), наполненную 1 г угля. Поглотительная трубка с двух сторон закрывается спиралью из проволоки во избежание высыпания угля. Продолжительность отбора не менее 50 мин.
б) Для определения среднесуточной концентрации воздух со скоростью до 1 л/мин. протягивают через трубку, наполненную 1 г угля. Отбор проб проводят непрерывно в течение суток. Допустимо отбор проб проводить в одну и ту же трубку 6 — 12 раз с перерывами в 4 или 2 ч.
Ход анализа
Уголь переносят в пробирку с притертой пробкой, добавляют 4 мл бензола. Содержимое пробирки встряхивают в течение 2 — 3 мин. и нагревают в течение часа при 55 — 60 °C, затем вновь встряхивают в течение 1 — 2 мин.
Для анализа берут 2 мл прозрачного раствора. В испытуемый раствор вносят 1 мл раствора ванадий-оксихинолинового комплекса в бензоле. Перемешивают и нагревают на водяной бане при 65 — 70 °C в течение 30 мин. После охлаждения добавляют 1 мл раствора едкого натра. Пробирку вновь интенсивно встряхивают до исчезновения черной окраски. Фотометрируют при длине волны 370 нм в кювете с толщиной слоя 1 см по сравнению с контролем. Содержание спиртов в анализируемом объеме определяют по калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов. Для этого рабочий стандартный расчет наносят на уголь, употребляемый для отбора проб, одновременно готовят контроль (табл. 59).
Таблица 59
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Бензол, мл |
2 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
|
Содержание спирта, мкг |
0 |
5,0 |
10 |
20 |
40 |
60 |
80 |
100 |
Каждую пробирку шкалы и контроль обрабатывают аналогично пробе. Измеряют оптическую плотность и строят калибровочный график.
Шкала может быть использована для визуального определения. При этом ее готовят одновременно с пробами в колориметрических пробирках.
Расчет см. выше.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ АЛИФАТИЧЕСКИХ СПИРТОВ
ГРУППЫ C1 — C10 ПРИ ИХ СОВМЕСТНОМ ПРИСУТСТВИИ С ПОМОЩЬЮ
БУМАЖНОЙ ХРОМАТОГРАФИИ [7]
Принцип определения
Спирты группы C1 — C10 переводят в соответствующие нелетучие производные 3,5-динитробензоата, которые разделяют затем на бумаге нисходящим методом в двух системах растворителей: диметилформамид-гексан (для C1 — C6) и вазелиновое масло — формамид (для C7 — C10).
После проявления хроматограммы производные спиртов обнаруживаются в виде желтых пятен. Количественное определение проводят фотометрически по изменению величины оптической плотности окрашенных элюатов, а также визуально сравнением интенсивности окраски проб с окраской «свидетелей».
Чувствительность по 0,5 мкг для каждого из спиртов C1 — C6 и 1 мкг для спиртов C6 — C10.
Определению не мешают алифатические кислоты группы C1 — C9, метиловые эфиры указанных кислот, амины C7 — C9, динил. Мешают определению соединения, содержащие гидроксильную группу.
Аппаратура
1. Теплоэлектровентилятор.
2. Пробирки с притертыми пробками емкостью 10 мл.
3. Хроматографические камеры размером 200 x 350 и 240 x 600 мм (рис. 26).
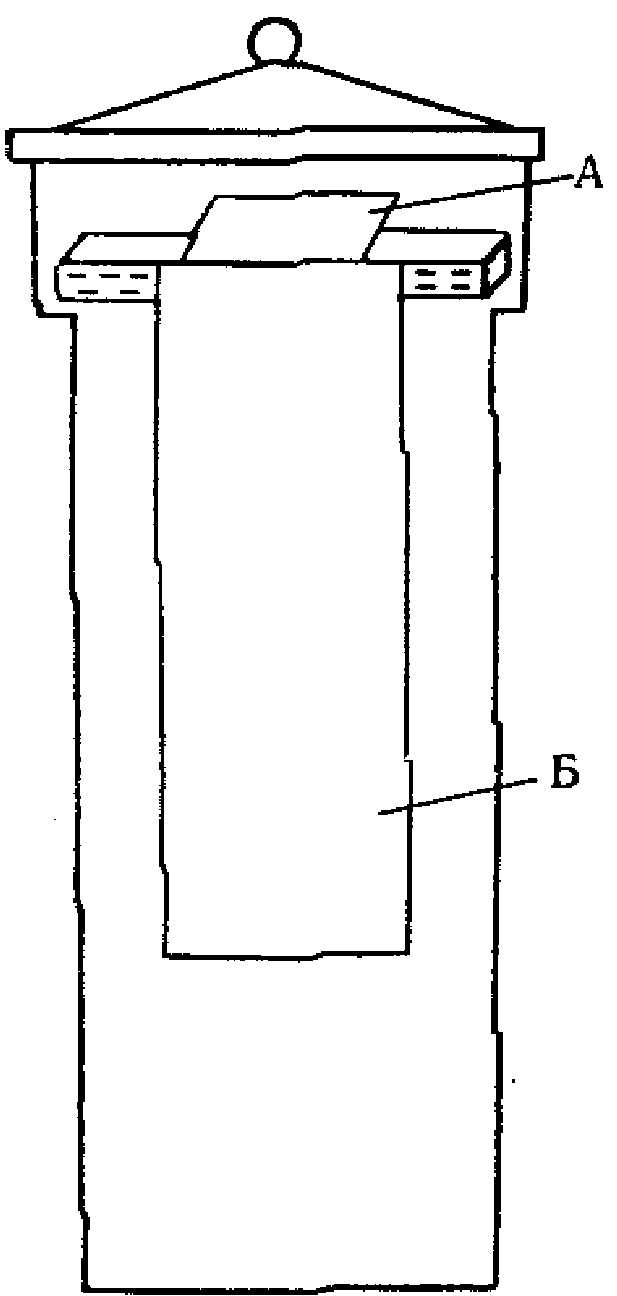
Рис. 26. Общий вид хроматографической камеры
А — лодочка с растворителем; Б — хроматографическая бумага
4. Делительные воронки емкостью 50 мл.
5. Эмалированная кювета размером 190 x 250 мм.
6. Мерные колбы емкостью 25 и 50 мл.
7. Микропипетки емкостью на 0,1 мл.
8. Резиновая груша.
9. Стеклянный распылитель.
10. Стеклянные трубки с четырьмя шариками для поглощения спиртов из воздуха (диаметр каждого шарика 10 мм).
11. Сушильный шкаф.
Реактивы
1. Активированный уголь марки АГ-5 обрабатывают концентрированной соляной кислотой в течение 1 — 1 1/2 ч при кипячении. Затем кислоту сливают, а уголь промывают несколько раз дистиллированной водой и 0,1 н. раствором аммиака до тех пор, пока последний не перестанет окрашиваться. Далее уголь снова промывают дистиллированной водой до отрицательной реакции на ион хлора и сушат тонким слоем при 100 — 120 °C в течение 1 — 2 ч. Хранят уголь в посуде с плотно прилегающей пробкой.
2. Стандартные растворы спиртов (метилового, этилового, пропилового, бутилового, амилового, гексилового, гептилового, октилового, нонилового, децилового) с содержанием 2 мг в 1 мл бензола.
3. 3,5-Динитробензоилхлорид — 10% раствор в бензоле (по весу).
4. Пиридин 10% раствор в бензоле (по объему).
5. Бензол.
6. Едкое кали 50% раствор.
7. Соляная кислота, 15% раствор.
8. Аммиак, 0,1% раствор.
9. Диметилформамид.
10. Гексан.
11. Вазелиновое масло.
12. Формамид.
13. Хлорид олова. Растворяют 0,7 г реагента в 5 мл воды, затем прибавляют 15 мл концентрированной соляной кислоты. Объем доводят до 100 мл водой. Раствор годен в течение суток.
14. П-Диметиламинобензальдегид. 1 г реагента растворяют в 95 мл этанола и прибавляют 5 мл концентрированной соляной кислоты. Раствор годен в течение 2 нед.
15. Метанол.
16. Ацетон.
17. Серная кислота, уд. вес 1,84.
18. Хроматографическая бумага марки «Ленинградская медленная» и «Ленинградская быстрая».
19. Этанол.
Отбор пробы
(см. выше «Определение общего количества алифатических спиртов» (C1 — C10).
Ход анализа
1. Получение 3,5-динитробензоатов спиртов
а) Обработка проб воздуха
После отбора пробы уголь переносят в пробирку с притертой пробкой и заливают 4 мл бензола. Содержимое пробирки встряхивают в течение 2 — 3 мин. и помещают пробирку на водяную баню, нагретую до 55 — 60 °C на один час. Затем пробирку вынимают из бани и снова встряхивают.
Для анализа берут 2 мл прозрачного раствора, переносят его в делительную воронку, куда добавляют 0,3 мл раствора 3,5-динитробензоилхлорида и 0,6 мл раствора пиридина. Объем пробы доводят до 5 мл бензолом, содержимое воронки встряхивают и оставляют на 30 мин. для образования 3,5-динитробензоатов спиртов.
Через 30 мин. производные спиртов промывают 5 мл раствора едкого кали, затем дважды водой по 25 мл каждый раз. Водный слой каждый раз удаляют, бензольный же промывают 5 мл 15% раствора соляной кислоты и дважды водой по 5 мл каждый раз.
Для получения более концентрированных проб бензол выпаривают на водяной бане. Полученный сухой остаток 3,5-динитробензоатов вновь растворяют в 0,15 мл бензола. 0,05 мл этого раствора используют для анализа спиртов группы C1 — C6 и 0,05 мл — для анализа спиртов C7 — C10.
б) Приготовление «свидетелей»
Стандартные растворы спиртов группы C1 — C6 смешивают в равных объемах по 0,5 мл, но не меньше, чем по 1 мг каждого. Так же поступают со стандартными растворами спиртов C7 — C10. Далее 3 мл смеси спиртов C1 — C6 (т.е. до 1 мг каждого спирта) помещают в делительную воронку. Дальнейшую обработку стандартных растворов спиртов проводят аналогично обработке проб воздуха.
Полученные растворы «свидетелей» содержат 3,5-динитробензоаты спиртов в количествах, соответствующих 0,4 мг/мл каждого из спиртов группы C1 — C10.
Производные спиртов могут сохраняться в течение 6 — 8 мес. при 6 — 8 °C.
2. Разделение 3,5-динитробензоатов
Для разделения производных спиртов C1 — C6 используется хроматографическая бумага марки «Ленинградская медленная». На листе размером 180 x 500 мм, отступя 7 см от края, проводят линию старта, на которой намечают четыре точки на расстоянии 3 — 3,5 см друг от друга.
С помощью микропипетки в одну точку наносят 0,05 мл раствора смеси соответствующих «свидетелей» спиртов группы C1 — C6, в три другие — пробы в количестве 0,05 мл. При этом диаметр пятен на линии старта не должен превышать 5 мм. С целью ускорения нанесения проб на бумагу можно использовать теплоэлектровентилятор. После нанесения пробы на бумагу последнюю «протягивают» через 50% раствор диметилформамида в ацетоне или этаноле, находящейся в эмалированной кювете. «Протягивают» бумагу от одного конца к другому до стартовой линии, не касаясь раствором проб, нанесенных на бумагу. Затем хроматографическую бумагу «отжимают» между листами фильтровальной бумаги и проделывают ту же операцию с другим ее концом. После этого бумагу подвешивают в течение 10 мин. для испарения растворителя, а затем помещают в хроматографическую камеру, предварительно насыщенную парами неподвижного и подвижного растворителей (диметилформамида и гексана).
Место соединения крышки с камерой заклеивают изоляционной лентой.
Разделение ведут нисходящим методом в течение 6 — 8 ч и считают законченным, когда подвижной растворитель — гексан — продвинется от линии старта на 40 см. После этого хроматограмму извлекают из камеры, сушат в течение 15 — 20 мин. на воздухе и проявляют.
Разделение производных спиртов групп C7 — C10 проводится на хроматографической бумаге марки «Ленинградская быстрая», которую после нанесения проб пропитывают 10% раствором вазелинового масла в гексане.
В качестве подвижной фазы используется формамид.
Разделение осуществляется в хроматографической камере размером 200 x 350 мм (см. рис. 26) и считается законченным, когда подвижной растворитель продвинется по бумаге на 25 см от линии старта. Далее хроматограмму сушат в вентилируемом сушильном шкафу при температуре 100 — 120 °C в течение 20 — 30 мин., а затем проявляют.
3. Обнаружение производных спиртов
Хроматограммы орошают раствором хлористого олова и через 1 — 2 ч раствором П-диметиламинобензальдегида. Производные спиртов C1 — C10 тотчас проявляются в виде круглых желтых пятен, окраска которых сохраняется более года.
Путем сравнения расположения на хроматограмме пятен проб с пятнами «свидетелей» делают выводы о наличии в пробах тех или иных спиртов, так как производные одного и того же спирта должны находиться на одинаковом расстоянии от линии старта.
Качественный состав пробы можно установить по величинам, которые вычисляют для каждого спирта отдельно (Rf — отношение расстояния пятна до линии старта к расстоянию от линии старта до линии фронта растворителя).
Затем, пользуясь табл. 60, находят отдельные спирты на хроматограмме.
Таблица 60
СРЕДНИЕ ВЕЛИЧИНЫ Rf ДЛЯ СПИРТОВ
|
Спирт |
Величина Rf в системе растворителей диметилформамид-гексан |
Спирт |
Величина Rf в системе растворителей вазелиновое масло-формамид |
|
CH3OH |
0,11 |
C7H13OH |
0,54 |
|
C2H5OH |
0,2 |
C8H17OH |
0,40 |
|
C3H7OH |
0,3 |
C9H19OH |
0,22 |
|
C4H9OH |
0,38 |
C10H21OH |
0,13 |
|
C5H11OH |
0,45 |
||
|
C6H13OH |
0,51 |
4. Количественное определение спиртов группы
C1 — C10 методом визуальной оценки
Готовят растворы «свидетелей» с различным содержанием производных спиртов. Для этого используют «свидетели», приготовленные согласно описанному выше способу. Бензол выпаривают, а сухие остатки, представляющие собой 3,5-динитробензоаты спиртов групп C1 — C6 и C7 — C10, растворяют каждый отдельно в 2,5 мл бензола (исходные растворы 1 и 2 соответственно).
Из полученных исходных растворов готовят серии разведений как показано в табл. 61 и 62.
Таблица 61
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ» ДЛЯ ОПРЕДЕЛЕНИЯ
СПИРТОВ ГРУПП C1 — C6
|
Исходный раствор спирта в бензоле, мл |
Бензол, мл |
Соответствующее количество каждого спирта, мкг |
|
|
в 1 мл |
в 0,05 мл |
||
|
2,5 |
0 |
400 |
20 |
|
0,8 |
0,2 |
320 |
16 |
|
0,6 |
0,4 |
240 |
12 |
|
0,4 |
0,6 |
160 |
8 |
|
0,2 |
0,8 |
80 |
4 |
|
0,05 |
0,95 |
20 |
1 |
Таблица 62
ПРИГОТОВЛЕНИЕ СЕРИИ СВИДЕТЕЛЕЙ ДЛЯ ОПРЕДЕЛЕНИЯ СПИРТОВ
ГРУППЫ C7 — C10
|
Исходный раствор спирта в бензоле, мл |
Бензол, мл |
Соответствующее количество каждого спирта, мкг |
|
|
в 1 мл |
в 0,05 мл |
||
|
2,5 |
0 |
400 |
20 |
|
0,6 |
0,4 |
240 |
12 |
|
0,4 |
0,6 |
160 |
8 |
|
0,2 |
0,8 |
80 |
4 |
|
0,05 |
0,95 |
20 |
1 |
Для определения спиртов на лист хроматографической бумаги в разные точки наносят по 0,05 мл раствора «свидетелей». Затем «свидетели» подвергаются хроматографическому анализу в тех же условиях, что и пробы.
На проявленных хроматограммах обнаруживаются пятна, разные по величине и интенсивности окраски и соответствующие 1 — 4 — 8 — 12 — 16 — 20 мкг каждого из спиртов групп C1 — C6 и 1 — 4 — 8 — 12 мкг — спиртов групп C7 — C10.
Количественное определение спиртов (после разделения их производных и проявления хроматограмм) методом визуальной оценки основано на том, что размер пятен и интенсивность окраски производных пропорциональны содержанию спиртов. Сравнивая интенсивность окраски пятен проб с окраской свидетелей, определяют содержание каждого спирта в пробе.
5. Количественное определение спиртов
групп C1 — C6 по оптической плотности элюатов
При фотометрическом определении содержания спиртов в пробе предварительно готовят стандартную шкалу, как описано выше, т.е. подвергают хроматографическому разделению «свидетели», соответствующие 1 — 4 — 8 — 12 — 16 — 20 мкг каждого из спиртов групп C1 — C6.
После проявления хроматограмм вырезают каждое пятно из бумаги, измельчают, помещают в пробирку и заливают 5 мл абсолютного метанола. Содержимое пробирки слегка встряхивают. Через 5 мин. окрашенный элюат переносят в кювету и измеряют величину его оптической плотности при длине волны 445 нм с помощью спектрофотометра или электрофотоколориметра.
Опыты повторяют 4 — 5 раз и по полученным средним данным строят градуировочные графики зависимости оптической плотности элюатов от концентрации вещества.
Такие графики строят для каждого спирта — метилового, этилового, пропилового, бутилового, амилового, гексилового.
Для определения содержания спиртов в пробах после их хроматографического анализа окраску каждого пятна элюируют, как описано выше, измеряют величину оптической плотности элюатов и с помощью графиков находят количество спиртов в пробе.
Расчет см. выше.
ИЗОПРОПИЛОВЫЙ СПИРТ (ИЗОПРОПАНОЛ)
Прозрачная бесцветная жидкость, температура кипения 82,8 °C, плотность при 20 °C 0,785, хорошо растворим в воде.
Общее действие на организм сходно с этанолом, но при равных концентрациях паров сильнее его.
Пары изопропанола раздражают слизистые оболочки глаз и верхних дыхательных путей, могут повредить сетчатку глаз и зрительный нерв.
Предельно допустимая концентрация: максимальная разовая 0,6 мг/м3.
ОПРЕДЕЛЕНИЕ ИЗОПРОПИЛОВОГО СПИРТА
С САЛИЦИЛОВЫМ АЛЬДЕГИДОМ [9]
Принцип определения
При взаимодействии изопропанола с персульфатом калия образуется ацетон, который с салициловым альдегидом в щелочной среде дает соединение, окрашивающее раствор от желтого до ярко-оранжевого цвета.
Чувствительность определения 2 мкг в анализируемом объеме раствора.
Ацетон мешает определению. Пропиловый спирт определению не мешает.
Реактивы
1. Поглотительный раствор — вода (дистиллированная).
2. Стандартный раствор. Готовят исходный раствор в мерной колбе емкостью 50 мл, наливают 5 мл воды и взвешивают. Затем в нее вносят 0,1 — 0,2 мл изопропанола и снова взвешивают. Раствор в колбе доводят до метки водой. По разности двух взвешиваний устанавливают содержание изопропанола в колбе и рассчитывают количество его в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержатся 500 мкг изопропанола. Раствор доводят до метки водой.
3. Изопропанол, температура кипения 82,8 °C.
4. Персульфат калия, 1% раствор.
5. Натр едкий, 40% раствор.
6. Альдегид салициловый, 20% раствор в этаноле.
7. Бисульфит калия, 5% раствор.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,3 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненные по 2 мл воды каждый. Продолжительность отбора 20 — 30 мин.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. В колориметрическую пробирку с пробкой переливают пробу (2 мл) и вносят в нее 0,2 мл персульфата калия. Пробирку быстро закрывают во избежание потери образовавшегося ацетона, ставят на водяную баню, которую постепенно нагревают до 50 — 54°, и при этой температуре оставляют пробирку на 30 мин. Затем пробирку вынимают и дают раствору охладиться при комнатной температуре. По охлаждении вносят 0,2 мл 5% раствора бисульфита и 2 мл 40% щелочи, содержимое пробирки встряхивают и вливают 0,2 мл салицилового альдегида. Раствор встряхивают и пробирку помещают в водяную баню, нагретую до 70 °C на 15 — 20 мин. По охлаждении пробу фотометрируют в кюветах с толщиной слоя 1 см при длине волны 445 — 450 нм по сравнению с контролем. Содержание изопропилового спирта в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 63). Шкалу стандартов можно использовать для визуального определения.
Таблица 63
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,8 |
1 |
1,5 |
2 |
|
Вода, мл |
2 |
1,8 |
1,6 |
1,2 |
1,0 |
0,5 |
0 |
|
Содержание изопропанола, мкг |
0 |
2 |
4 |
8 |
10 |
15 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
ЦИКЛОГЕКСАНОЛ
Бесцветная жидкость, обладающая запахом камфоры, температура кипения 161 °C (технического 155 — 165), плотность 0,962 при 20 °C, растворима в воде.
Циклогексанол раздражающе действует на слизистую оболочку глаз.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,06 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИКЛОГЕКСАНОЛА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [6]
Принцип определения
При взаимодействии циклогексанола с парадиметиламинобензальдегидом в сернокислой среде образуется соединение, окрашивающее раствор в розовый цвет.
Чувствительность определения 0,8 мкг в анализируемом объеме раствора.
Амины, высшие спирты мешают определению.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор циклогексанола: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 5 — 10 мл воды, и взвешивают. В колбу вносят 2 — 3 капли циклогексанола и снова взвешивают. По разности двух взвешиваний устанавливают количество циклогексанола. Объем жидкости в колбе доводят до метки водой. Вычисляют количество циклогексанола в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг циклогексанола. Раствор доводят до метки водой.
3. Серная кислота, уд. вес 1,84 и 1,727, т.е. 80%.
4. Парадиметиламинобензальдегид, 0,5% раствор в 80% серной кислоте.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 3 мл дистиллированной воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
В пробирку помещают 2 мл пробы, в которую наливают 0,2 мл парадиметиламинобензальдегида и затем 4 мл серной кислоты удельного веса 1,84. Содержимое пробирки встряхивают и помещают на 10 мин. в кипящую водяную баню. По охлаждении раствора измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 510 нм.
Содержание циклогексанола в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 64).
Таблица 64
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,08 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода, мл |
2 |
1,92 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание циклогексанола, мкг |
0 |
0,8 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалу стандартов можно использовать для визуального определения, шкалу готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
ФЕНОЛ
(Оксибензол, карболовая кислота)
Бесцветные кристаллы, краснеющие на воздухе.
Температура плавления 41 °C, температура кипения 182 °C, растворяется в воде 6,7 г в 100 мл при температуре 16 °C, при 66 °C смешивается с водой во всех отношениях.
Растворим в этаноле, эфире, хлороформе и других органических растворителях.
При вдыхании паров фенола наблюдаются раздражение дыхательных путей, расстройство пищеварения, общая и мышечная слабость и другие патологические явления.
При попадании фенола на кожу в местах соприкосновения с фенолом происходит омертвение тканей, общее отравление организма, нарушаются функции кровообращения, дыхания, повышается температура.
Предельно допустимые концентрации: максимальная разовая 0,01 мг/м3, среднесуточная 0,01 мг/м3.
ОПРЕДЕЛЕНИЕ ФЕНОЛА С П-НИТРОФЕНИЛДИАЗОНИЕМ [4]
Принцип определения
При взаимодействии фенола с п-нитрофенилдиазонием в щелочной среде образуется продукт, окрашенный в красный цвет.
Чувствительность определения 0,5 мкг в определяемом объеме раствора.
Определению мешают орто-, метакрезолы, амины.
Реактивы
1. Поглотительный раствор — 0,8% раствор карбоната натрия.
2. Силикагель КСМ, КСК, величина зерен 0,5 — 2 мм (предварительно обработанный) (см. ниже).
3. Стандартный раствор фенола. Исходный раствор с содержанием (приблизительно) 0,5 мг фенола в 1 мл готовят растворением около 0,125 г фенола в мерной колбе емкостью 250 мл. Доводят до метки 0,8% раствором карбоната натрия. Точное содержание вычисляют по навеске.
Рабочий стандартный раствор с содержанием 10 мкг в 1 мл готовят соответствующим разбавлением исходного.
Рабочие растворы используют свежеприготовленными.
4. Нитрит натрия, 25% раствор (свежеприготовленный).
5. Паранитроанилин.
6. Соляная кислота, уд. вес 1,19.
7. П-Нитрофенилдиазоний. Готовят добавлением к 50 мл воды 2,5 мл соляной кислоты уд. веса 1,19, 2,5 мл 25% раствора нитрита натрия и 0,01 г паранитроанилина. Тщательно перемешивают. Раствор применяется свежеприготовленным.
8. Этанол.
Отбор пробы
а) Для определения разовой концентрации отбирают пробу воздуха в «кипящий» слой силикагеля. Для этого по 3 мл силикагеля вносят через воронку в два поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана, соединенные последовательно, к которым присоединен поглотитель для улавливания переброшенных частиц силикагеля. Воздух протягивают со скоростью до 6 л/мин. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 3 л/мин. протягивают в «кипящий» слой силикагеля в течение суток непрерывно.
Допустимо проводить отбор в одну и ту же систему поглотителей 6 — 12 раз с перерывом в 2 — 4 ч.
При определении концентраций фенола, превышающих допустимые, воздух со скоростью 0,5 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористым фильтром N 1, наполненных по 6 мл поглотительного раствора.
Ход анализа
а) При отборе проб на силикагель последний из каждого поглотительного прибора отдельно переносят в пробирки с притертыми пробками. Переброшенные зерна силикагеля (из последнего прибора) присоединяют к силикагелю из второго поглотительного прибора.
Добавляют по 2 мл этанола, перемешивают и через 30 мин. добавляют по 2 мл поглотительного раствора. Вновь взбалтывают и дают отстояться. Для анализа берут по 2 мл прозрачного раствора.
Одновременно готовят шкалу стандартов и контроль. Для этого в пробирки помещают по 3 мл силикагеля, употребляемого при отборе, наносят на силикагель стандартные растворы фенола и другие реактивы, согласно табл. 65.
Таблица 65
ШКАЛА СТАНДАРТОВ ПРИ ОТБОРЕ ПРОБ НА СИЛИКАГЕЛЬ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Этанол, мл |
По 1 мл во все пробирки |
||||||
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0,0 |
|
Содержание фенола, мкг |
0 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Стандартную шкалу обрабатывают аналогично пробам.
Содержание фенола устанавливают путем визуального сравнения интенсивности окраски пробы со шкалой.
б) При отборе проб в жидкую поглотительную среду содержимое каждого поглотительного прибора переносят в пробирки. Для анализа берут 5 мл пробы. Добавляют по 0,2 мл раствора п-нитрофенилдиазония. Содержимое пробирки встряхивают и через 10 мин. фотометрируют при длине волны 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем.
Содержание фенола в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 66).
Таблица 66
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,5 |
|
Содержание фенола, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
15,0 |
В каждую пробирку шкалы стандартов добавляют по 0,2 мл раствора п-нитрофенилдиазония, перемешивают, измеряют оптическую плотность и строят калибровочный график.
Шкала стандартов может быть использована для визуального определения. В этом случае ее готовят в колориметрических пробирках одновременно с пробами.
ОПРЕДЕЛЕНИЕ ФЕНОЛА С 4-АМИНОАНТИПИРИНОМ [10]
Принцип определения
При взаимодействии фенола с 4-аминоантипирином в присутствии железосинеродистого калия при pH среды 9,3 образуется продукт, окрашенный в красный цвет.
Чувствительность — 0,5 мкг в определяемом объеме раствора.
Мешает определению ацетофенон в количествах, превышающих 20 мкг. Не мешают определению паракрезол, бензол, изопропилбензол, гидроперекись изопропилбензола, ацетон, стирол, ![]() -метилстирол, диметилвинилпараоксифенилметан.
-метилстирол, диметилвинилпараоксифенилметан.
Реактивы
1. Поглотительный раствор — тетраборат натрия Na2B4O7 ·10H2O, 0,05 М раствор. Готовят растворением 1 г тетрабората в 1 л воды.
2. Силикагель КСК, КСМ, величина зерен 0,5 — 2 мм (предварительно обработанный, см. ниже).
3. Стандартные растворы фенола с содержанием 500; 10 и 1 мкг/мл готовят так же, как указано в методе с п-нитрофенилдиазонием.
4. 4-Аминоантипирин, 1% раствор.
5. Железосинеродистый калий (K3[Fe(CN)6]), 0,1% раствор.
6. Этанол.
Отбор проб
См. «Определение фенола с п-нитрофенилдиазонием».
Ход анализа
а) При отборе проб в жидкую среду содержимое каждого поглотительного прибора переносят в пробирки. Для анализа берут 5 мл пробы. Добавляют по 0,1 мл 1% раствора 4-аминоантипирина, по 0,1 мл 0,1% раствора железосинеродистого калия.
Содержимое пробирок перемешивают и через 10 мин. фотометрируют при длине волны 495 нм в кювете с толщиной слоя 1 см по сравнению с контролем. Содержание фенола в анализируемом объеме определяют по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 67).
Таблица 67
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,7 |
4,6 |
4,5 |
4,4 |
|
Содержание фенола, мкг |
0 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
6,0 |
В каждую пробирку шкалы вносят по 0,1 мл 1% раствора 4-аминоантипирина и поступают аналогично анализу пробы.
Шкала стандартов может быть использована для визуального определения. Ее в этом случае готовят одновременно с пробами в колориметрических пробирках.
б) При отборе проб на силикагель поступают так же, как и при определении фенола с п-нитрофенилдиазонием.
После добавления к силикагелю этилового спирта вносят в пробу по 2 мл раствора тетрабората натрия и далее все реактивы в том же порядке, как при определении фенола, поглощенного в жидкую среду.
Интенсивность окраски проб визуально сравнивают с интенсивностью окраски шкалы (табл. 67).
Расчет см. выше.
СЛОЖНЫЕ ЭФИРЫ
RCOOR1
Сложные эфиры уксусной кислоты — бесцветные летучие жидкости, обладающие характерным запахом. Растворимость их в воде ограничена и уменьшается с увеличением молекулярного веса. Легко воспламеняются, в определенной смеси с воздухом взрывоопасны. Относятся к токсическим веществам, их пары раздражают слизистые оболочки дыхательных путей, глаз.
Таблица 68
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА И ПРЕДЕЛЬНО ДОПУСТИМЫЕ
КОНЦЕНТРАЦИИ (ПДК) ЭФИРОВ УКСУСНОЙ КИСЛОТЫ
|
Название |
Формула |
Молекулярный вес |
Плотность |
Температура кипения при 760 мм рт. ст. |
ПДК мг/м3 |
|
|
максимальная разовая |
среднесуточная |
|||||
|
Амилацетат |
CH3COOC2H5 |
130,19 |
0,879 (20°) |
142,5 |
0,1 |
0,1 |
|
Бутилацетат |
CH3COOC4H9 |
116,16 |
0,883 (20°) |
125 (740 мм рт. ст.) |
0,1 |
0,1 |
|
Винилацетат |
CH3COOCH2CH2 |
86,09 |
0,932 (20°) |
72,3 |
0,3 |
0,2 |
|
Метилацетат |
CH3COOCH3 |
74,08 |
0,934 |
57,3 |
0,07 |
0,07 |
|
Этилацетат |
CH3COOC2H5 |
88,10 |
0,900 |
77,15 |
0,1 |
0,1 |
ОПРЕДЕЛЕНИЕ СЛОЖНЫХ ЭФИРОВ (АМИЛАЦЕТАТА, БУТИЛАЦЕТАТА,
ВИНИЛАЦЕТАТА, МЕТИЛАЦЕТАТА, ЭТИЛАЦЕТАТА)
С СОЛЯНОКИСЛЫМ ГИДРОКСИЛАМИНОМ [5]
Принцип определения
При взаимодействии сложных эфиров с солянокислым гидроксиламином в щелочной среде образуются гидроксаматы, которые реагируют с хлорным железом, давая продукт, окрашенный в фиолетовый цвет.
Чувствительность определения 5 мкг в анализируемом объеме раствора.
Ангидриды кислот, сложные эфиры адипиновой, акриловой, малеиновой кислот мешают определению.
Реактивы
1. Силикагель АСМ, КСМ, КСК с величиной зерен 0,25 — 1 мм, обработанный соляной кислотой, разбавленной водой в отношении 1:1 при нагревании в течение 3 — 4 ч, промывают водой. После этого силикагель высушивают при 100 — 105 °C и активируют при 200 °C в течение 30 мин.
Регенерация силикагеля проводится тем же способом.
2. Этанол, предварительно обработанный едкой щелочью. Для этого в колбу емкостью 500 мл всыпают 20 — 30 г едкого натра, вливают этанол, раствор встряхивают и оставляют на 2 — 3 ч, временами встряхивая его. После этого раствор перегоняют при 78 °C.
3. Стандартные растворы сложных эфиров готовят в этаноле. В мерную колбу емкостью 25 — 30 мл вливают 10 — 15 мл этанола, взвешивают, вносят в колбу эфир, который предполагают определять, 4 — 5 капель, и снова взвешивают. По разности двух взвешиваний определяют количество эфира и вычисляют его содержание в 1 мл раствора. Объем раствора доводят до метки этанолом. Рабочий стандартный раствор с содержанием 100 мкг/мл готовят в мерной колбе емкостью 50 мл, в которую вливают количество исходного раствора с содержанием 5000 мкг эфира. Объем раствора доводят до метки этанолом.
4. Гидроксиламин солянокислый, 4 н. раствор.
5. Едкий натр, 4 н. раствор.
6. Соляная кислота, 0,5 н. и 4 н. раствор.
7. Хлорное железо окисное, 1,6% раствор в 0,5 н. растворе соляной кислоты.
8. Фенолфталеин, 0,5% спиртовой раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 5 — 10 л/мин. протягивают в «кипящий» слой силикагеля. Для этого в два последовательно присоединенных поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана или поглотителя Яворовской помещают по 3 мл силикагеля, к ним присоединяют прибор для улавливания переброшенных частиц силикагеля.
Винилацетат поглощают на неподвижный слой силикагеля, помещенный в шариковую трубку.
Продолжительность отбора 20 — 30 мин.
б) При определении среднесуточной концентрации отбор проб проводят в «кипящий» слой силикагеля со скоростью 1 — 2 л/мин. в течение суток непрерывно. Допустимо отбор проводить 6 — 12 раз с перерывами в 4 — 2 ч по 20 — 30 мин.
Ход анализа
Силикагель переносят из каждого поглотительного прибора отдельно в пробирки с притертыми пробками, добавляют по 6 мл этанола и помещают пробирки на водяную баню, нагретую до 50 °C на 20 мин. Содержимое пробирок периодически встряхивают. Раствору дают отстояться. Для определения берут по 3 мл прозрачного раствора, в пробирку вливают 0,2 мл 4 н. раствора гидроксиламина, 0,6 мл 4 н. раствора едкого натра. Раствор встряхивают и оставляют на 15 мин. Затем раствор нейтрализуют 4 н. соляной кислотой по фенолфталеину и в пробирку вливают 2,5 мл 1,6% раствора хлорида железа. Раствор встряхивают и через 20 — 30 мин. переливают в кювету толщиной слоя 1 см и измеряют оптическую плотность раствора, сравнивая с контролем, который готовят одновременно и аналогично пробе. Определение проводят с помощью фотоэлектроколориметра при длине волны 500 нм.
Содержание эфира в анализируемом объеме определяют по градуировочному графику, который предварительно строят по шкале стандартов (табл. 69).
Таблица 69
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,10 |
0,20 |
0,30 |
0,40 |
0,5 |
|
Этанол, мл |
1,5 |
1,45 |
1,40 |
1,30 |
1,20 |
1,10 |
1,0 |
|
Содержание эфира, мкг |
0 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалу стандартов можно использовать для визуального определения, готовят ее одновременно с пробами в колориметрических пробирках. Первая пробирка содержит 2 мкг эфира.
Расчет см. выше.
Примечание. При концентрации эфиров выше допустимой нормы отбор проб можно проводить в спирт со скоростью 0,3 — 0,5 л/мин.
ОПРЕДЕЛЕНИЕ ВИНИЛАЦЕТАТА ХРОМАТОГРАФИЕЙ НА БУМАГЕ [11]
Принцип определения
При взаимодействии винилацетата с ацетатом ртути образуется нелетучее ртутноорганическое соединение. Его выделяют на бумаге в системе растворителей бутанол — триэтиламин — вода нисходящим способом. Проявляющим реактивом служит раствор дифенилкарбазида.
Чувствительность 3 мкг в пробе. Определению не мешают 2-этилгексилакрилат, винилхлорид, дибутилмалеинат, диоктилфталат, дибутилфталат, стирол, ![]() -метилстирол.
-метилстирол.
Аппаратура
1. Электровентилятор.
2. Хроматографическая камера размером 260 x 600 мм с лодочкой и подставкой для лодочки (см. рис. 26).
3. Микропипетки емкостью 0,1 мл с делениями на 0,01 мл.
4. Стеклянный распылитель (пульверизатор).
Реактивы
1. Винилацетат, перегнанный при 72,5 °C. Исходный стандартный раствор готовят в мерной колбе емкостью 10 мл, в которую вносят 2 — 3 мл поглотительного раствора, колбу взвешивают и добавляют 1 — 2 капли винилацетата. Раствор в колбе доводят до метки поглотительным раствором. Рассчитывают содержание винилацетата в 1 мл раствора. Соответствующим разбавлением готовят раствор N 1 с содержанием 200 мкг/мл. Рабочие стандартные растворы готовят из раствора N 1 в соответствии с табл. 70.
Таблица 70
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Стандартный раствор N 1, мл |
0 |
0,45 |
0,60 |
0,75 |
1,05 |
1,5 |
|
Поглотительный раствор, мл |
3 |
2,55 |
2,40 |
2,25 |
1,95 |
1,6 |
|
Содержание винилацетата в 1 мл раствора, мкг |
0 |
30 |
40 |
50 |
70 |
100 |
2. Поглотительный раствор: к 100 мл пропилового спирта добавляют 0,5 мл ледяной уксусной кислоты и 0,2 г ацетата ртути.
3. Пропиловый спирт.
4. Бутиловый спирт.
5. Ацетат ртути, двузамещенная соль.
6. Кислота уксусная, ледяная.
7. Триэтиламин.
8. Дифенилкарбазид, 0,15% раствор.
9. Этиловый спирт очищенный. К 1 л спирта добавляют 40 г едкого кали, оставляют на 4 ч, затем отгоняют при 78,8 °C.
10. Система растворителей. В делительную воронку вносят 25 мл бутанола, 7 мл триэтиламина и 27 мл воды. Смесь встряхивают в течение 10 мин. и оставляют до полного расслоения.
11. Бумага хроматографическая «Медленная».
Отбор пробы
Воздух со скоростью 1 л/мин. протягивают через две пары параллельно соединенных поглотительных приборов с пористой пластинкой N 2, содержащих по 3 мл поглотительного раствора.
Отбор проводят в течение 20 — 30 мин. при охлаждении.
Ход анализа
Жидкость из первых двух поглотительных приборов переносят в центрифужные пробирки с делениями. Также объединяют и поглотительные жидкости из вторых поглотительных приборов.
Пробирки помещают в кипящую водяную баню и выпаривают спирт до объема 0,3 мл (по метке).
В хроматографической камере по стенкам располагают фильтровальную бумагу и смачивают ее нижним слоем, полученным при разделении слоев жидкостей системы, предназначенной для разделения смеси. Верхний слой жидкости переносят из делительной воронки в лодочку. Насыщение хроматографической камеры парами растворителей проводят в течение 4 — 5 ч.
На полоске хроматографической бумаги на расстоянии 6 см от края намечают линию старта, на которой отмечают три точки на расстоянии 2,5 см друг от друга, в которые при помощи микропипетки дробно наносят пробу, обдувая теплым воздухом. Затем пробирку обмывают 0,1 — 0,15 мл пропилового спирта и смыв наносят в те же точки. Хроматографическую бумагу с нанесенной пробой помещают в лодочку, находящуюся в хроматографической камере. Камеру герметизируют и оставляют на 15 — 17 ч при комнатной температуре. При этом растворитель опускается по бумаге на расстояние около 25 см.
Далее бумагу извлекают из камеры, высушивают при комнатной температуре, орошают раствором дифенилкарбазида и термостатируют в течение 3 мин. при 60 — 70 °C. При наличии винилацетата на хроматограмме проявляются зоны локализации в виде круглых пятен, окрашенных в фиолетовый цвет, с величиной Rf 0,7. На линии старта также проявляются окрашенные зоны, свидетельствующие об избытке ацетата ртути.
Зоны, характерные для винилацетата, вырезают из бумаги, измельчают и помещают в пробирку, в которую вносят 4 мл этилового спирта. Содержимое пробирки встряхивают трижды в течение 15 — 20 мин., после чего раствор центрифугируют.
Оптическую плотность измеряют на спектрофотометре в кюветах с толщиной слоя 1 см при длине волны 560 нм.
Содержание винилацетата устанавливают по калибровочному графику. Для построения калибровочного графика готовят несколько стандартных шкал.
Согласно шкалы стандартов (табл. 70), получают ряд растворов — N 1 — 2 — 3 и т.д. с содержанием от 30 до 100 мкг винилацетата в 1 мл.
На другой лист хроматографической бумаги по линии старта в отдельные точки наносят по 0,1 мл указанных в табл. 70 растворов, что соответствует N 3 — 4 — 5 — 7 и 10 мкг винилацетата.
Одновременно готовят контроль. При этом в контрольную точку наносят 0,1 мл поглотительного раствора. Бумагу с нанесенной стандартной шкалой помещают в ту же лодочку, в которую помещена проба, располагая ее по другую сторону хроматографической камеры. При этом достигают идентичности условий хроматографирования.
После хроматографирования шкалу орошают раствором дифенилкарбазида, производят те же операции и измеряют величины оптических плотностей, после чего строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения.
Расчет см. выше.
МЕТИЛОВЫЙ ЭФИР МЕТАКРИЛОВОЙ КИСЛОТЫ
(метилметакрилат)
|
CH2=C(CH3)COOCH3 |
Мол. вес 100,11 |
Прозрачная жидкость, температура кипения 100 °C, плотность при 15,6 °C 0,95, нерастворим в воде, хорошо растворим в органических растворителях. Легко полимеризуется; гидрохинон, пирогаллол задерживают полимеризацию.
Обладает наркотическим и общетоксическим действием.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,1 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТИЛМЕТАКРИЛАТА ПО ФОРМАЛЬДЕГИДУ [1]
Принцип определения
При омылении метилметакрилата щелочью образуется метанол, который окисляют до формальдегида. Последний определяют с хромотроповой кислотой.
Чувствительность определения — 2 мкг в анализируемом объеме раствора.
Метанол, формальдегид, метиловые эфиры других кислот мешают определению.
Реактивы
1. Поглотительный раствор — 2,5% раствор едкого натра.
2. Стандартный раствор. Готовят исходный раствор в мерной колбе емкостью 50 мл, вливают 5 — 10 мл 25% раствора едкого натра, колбу взвешивают и вносят 2 — 3 капли метилметакрилата, снова взвешивают и объем в колбе доводят до метки 2,5% раствором едкого натра. По разности двух взвешиваний устанавливают количество метилметакрилата в колбе и вычисляют содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в другой колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг метилметакрилата. Объем раствора доводят до метки 2,5% раствором едкого натра.
3. Едкий натр, 2,5% раствор.
4. Серная кислота, разбавленная водой в отношении 1:1.
5. Перманганат калия, 2% раствор.
6. Сульфит натрия, 30% раствор, свежеприготовленный.
7. Хромотроповая кислота или ее динатриевая соль; 0,1 г этой кислоты растворяют в 2 мл воды и добавляют до 50 мл 15 М серной кислотой.
8. 15 М серная кислота. К 10 мл воды осторожно приливают при охлаждении 90 мл серной кислоты уд. веса 1,82 — 1,84.
9. Серная кислота, уд. вес 1,82 — 1,84.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненных по 4 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,3 л/мин. протягивают через указанные выше приборы, содержащие по 4 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в одни и те же приборы по 20 — 30 мин.
Ход анализа
Анализируют каждый поглотительный прибор отдельно. Берут 2 мл пробы в пробирку, вливают 0,5 мл серной кислоты, разбавленной водой в отношении 1:1, и по капле раствора перманганата калия. Растворы взбалтывают и через 5 — 10 мин. вносят по одной капле сульфита для обесцвечивания раствора. Избытка сульфита необходимо избегать.
Затем вливают 2 мл хромотроповой кислоты. Раствор встряхивают и помещают на 5 мин. в кипящую водяную баню.
Раствор охлаждают и измеряют оптическую плотность раствора в кювете шириной 1 см по сравнению с контролем при длине волны 570 нм. Содержание метилметакрилата в анализируемом объеме определяют по предварительно построенному градуировочному графику.
Для построения графика готовят шкалу стандартов (табл. 71).
Таблица 71
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
0,5 |
— |
|
Содержание метилметакрилата, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
15 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалу стандартов можно использовать для визуального определения, ее следует готовить в колориметрических пробирках одновременно с пробой, шкалу начинать с 1 мкг и далее, как указано в табл. 71.
Расчет см. выше.
МЕТИЛОВЫЙ ЭФИР АКРИЛОВОЙ КИСЛОТЫ
(Метилакрилат)
|
CH2=CH-COO·CH3 |
Мол. вес 86,09 |
Прозрачная бесцветная жидкость с резким запахом, температура кипения 80 °C, плотность при 20 °C 0,95, незначительно растворим в воде, но хорошо растворим в органических растворителях, легко полимеризуется. Обладает наркотическим и общетоксическим действием на организм, а также резко раздражающим действием.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТИЛАКРИЛАТА ПО ФОРМАЛЬДЕГИДУ [1]
Определение метилакрилата проводится так же, как метилметакрилата. Все реактивы те же, кроме стандартного раствора, его готовят из метилакрилата (см. выше).
ТЕТРАГИДРОФУРАН (ТГФ)
(Окись диэтилена)
|
|
Мол. вес 72,06 |
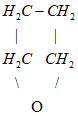
Жидкость с запахом этилового эфира, удельный вес ее при 20 °C 0,889, температура кипения 64 — 66 °C, смешивается с водой и спиртом в любых отношениях. Упругость пара ТГФ при 20 °C составляет 131,50, испаряется в 2,8 раза медленнее этилового эфира. ТГФ в промышленности применяется как растворитель и инсектицид. Тетрагидрофуран обладает наркотическим действием, раздражающим действием на слизистые оболочки глаз и верхние дыхательные пути, а также вызывает поражение почек.
Предельно допустимые концентрации ТГФ: максимальная разовая 0,2 мг/м3, среднесуточная 0,2 мг/м3.
ОПРЕДЕЛЕНИЕ ТЕТРАГИДРОФУРАНА
С ПАРАДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [3]
Принцип определения
При взаимодействии ТГФ с п-диметиламинобензальдегидом в сернокислой среде образуется окрашенный в розовый цвет продукт конденсации.
Чувствительность определения — 5 мкг ТГФ в пробе. Определению мешают фуран, спирты, а также соединения, дающие бурую окраску для реакции с серной кислотой. Фурфурол определению не мешает.
Реактивы
1. Силикагель марки АСМ или КСМ с зернами размером 0,3 — 0,5 мм.
Обработку силикагеля см. выше. Обработанный силикагель не должен давать реакцию на ТГФ.
2. Стандартные растворы ТГФ.
Исходный стандартный раствор готовят в мерной колбе емкостью 50 мл, куда вносят 10 мл воды, взвешивают и вносят несколько капель ТГФ, вновь взвешивают, доводят до метки и по разности весов устанавливают содержание ТГФ в 1 мл.
Соответствующим разбавлением готовят рабочий стандартный раствор с содержанием 100 мкг ТГФ в 1 мл.
3. 2% спиртовой раствор парадиметиламинобензальдегида.
4. Спирт, 96° ректификат.
5. Серная кислота концентрированная.
6. Соляная кислота, 1:2.
Отбор пробы
Для определения разовой концентрации в два последовательно соединенных поглотительных прибора НИИ гигиены имени Ф.Ф. Эрисмана засыпают по 3 мл силикагеля и протягивают анализируемый воздух со скоростью 5 — 10 л/мин. в кипящий слой силикагеля. Для улавливания переброшенных частиц силикагеля после второго поглотительного прибора ставят ловушку. Переброшенный силикагель присоединяют к силикагелю из второго поглотительного прибора. Отбор проводят в течение 20 — 30 мин.
Ход анализа
Силикагель переносят в две пробирки с притертыми пробками и заливают 2 мл дистиллированной воды и 8 мл концентрированной серной кислоты. Добавляют по 0,2 мл 2% раствора п-диметиламинобензальдегида. Содержимое пробирок осторожно встряхивают и выдерживают в кипящей водяной бане в течение часа.
В отдельной пробирке такой же обработке подвергается силикагель, не содержащий ТГФ (контроль).
По охлаждении пробирок окрашенный раствор над силикагелем сливают в кюветы с толщиной слоя 2 см и пробы фотометрируют при длине волны 520 нм.
Содержание ТГФ в пробе определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 72), при этом стандартный раствор наносят на 3 мл силикагеля, помещенного в пробирки.
Таблица 72
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Вода, мл |
2 |
1,95 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
|
Серная кислота, мл |
По 8 мл |
||||||
|
Содержание ТГФ, мл |
0 |
5 |
10 |
20 |
40 |
60 |
80 |
Все пробирки шкалы обрабатывают аналогично пробам, измеряют оптическую плотность и строят график.
Шкалой стандартов можно пользоваться и для визуального определения, при этом растворы проб и шкалы стандартов можно не сливать с силикагеля.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение метилметакрилата. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеева М.В. Определение метилового спирта. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
3. Деяпова Е.В. Определение тетрагидрофурана. В кн.: Методы определения вредных веществ в воздухе. М., «Медицина», 1966.
4. Ефремова Г.П. Определение фенола. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., «Медицина», 1955, в. 2.
5. Кузьмичева М.Н. Определение сложных эфиров в воздухе. Гиг. труда и профзаболеваний, 1961, 5.
6. Масленников А.С. Определение малых количеств циклогексанола в водных растворах и в воздухе. Заводская лаборатория, 1954, 1.
7. Пинигина И.А. Раздельное определение алифатических спиртов в воздухе с помощью хроматографии на бумаге. Гиг. и сан., 1965, 11.
8. Пинигина И.А. О колориметрическом определении паров спиртов в воздухе. Гиг. и сан., 1966, 1.
9. Селина И.А. Определение вторичного пропилового (изопропилового) спирта в воздухе. Гиг. и сан., 1962, 12.
10. Хрусталева В.А. Определение фенола в атмосферном воздухе. Гиг. и сан., 1962, 10.
11. Хрусталева В.А., Осокина С.К. Раздельное определение винилацетата и 2-этилгексилакрилата в присутствии дибутилмалеината в воздухе. Гиг. и сан., 1970, 4.
Глава XI
АЛЬДЕГИДЫ, КЕТОНЫ
ФОРМАЛЬДЕГИД
Бесцветный газ с резким запахом, хорошо растворимый в воде; 35 — 40% раствор формальдегида называется формалином.
Температура кипения формальдегида — 21 °C.
Легко полимеризуется, образуя параформальдегид.
Оказывает раздражающее действие на верхние дыхательные пути и глаза. При попадании растворов формалина на кожу могут возникать дерматиты. Формальдегид обладает и общей токсичностью.
Предельно допустимые концентрации: максимальная разовая 0,035 мг/м3; среднесуточная 0,035 мг/м3.
ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА С ХРОМОТРОПОВОЙ КИСЛОТОЙ [1, 6]
Принцип определения
При взаимодействии формальдегида с хромотроповой кислотой образуется соединение, окрашенное в фиолетовый цвет. Интенсивность окраски пропорциональна количеству формальдегида.
Чувствительность определения при фотометрическом определении — 0,2 мкг в анализируемом объеме раствора.
Определению мешают: сероводород с 0,03 мг/м3, двуокись азота и метиловый спирт с 0,3 мг/м3, сернистый газ с 3 мг/м3, аммиак с 0,5 мг/м3, ацетон с 6 мг/м3.
Реактивы
1. Силикагель КСК, КСМ, промытый соляной кислотой, водой до нейтральной реакции, высушенный и активированный при 200 °C в течение 30 мин. Регенерация силикагеля осуществляется тем же способом.
2. Стандартный раствор формальдегида. 5 мл водного раствора формальдегида (40% раствор формалина) доводят в мерной колбе емкостью 250 мл до метки водой и определяют содержание формальдегида в этом растворе. Для этого 5 мл раствора помещают в коническую колбу емкостью 200 мл с притертой пробкой, приливают 20 мл 0,1 н. раствора йода и по каплям вносят 30% раствор едкого натра до появления устойчивой бледно-желтой окраски. Колбу оставляют на 10 мин., затем осторожно подкисляют раствор 2,5 мл соляной кислоты (1:5), оставляют на 10 мин. в темноте и оттитровывают избыток йода 0,1 н. раствором тиосульфата натрия. Когда раствор станет светло-желтым, прибавляют несколько капель 0,5% раствора крахмала. Предварительно устанавливают количество тиосульфата, расходуемое на титрование 20 мл 0,1 н. раствора. По разности между количеством тиосульфата, израсходованным на контрольное титрование, и избытком йода, не вошедшим в реакцию с формальдегидом, устанавливают количество йода, которое пошло на окисление формальдегида, 1 мл 0,1 н. раствора йода соответствует 1,5 мг формальдегида.
Установив содержание формальдегида в 1 мл раствора, соответствующим разведением водой готовят исходный стандартный раствор формальдегида с содержанием 100 мкг/мл. Раствор проверяют титрометрически.
Рабочий стандартный раствор с содержанием 1 мкг/мл формальдегида готовят в день анализа разбавлением исходного стандартного раствора в 100 раз дистиллированной водой.
3. Соляная кислота, приготовленная в отношении 1:5.
4. Йод, 0,1 н. раствор.
5. Тиосульфат, 0,1 н. раствор.
6. Серная кислота, 9 М раствор. К 530 мл воды осторожно приливают 470 мл серной кислоты удельного веса 1,80 — 1,82.
7. Хромотроповая кислота (1,8-диоксинафталин-3,6-дисульфокислота), 0,5 г кислоты растворяют в 10 мл воды. Хранят раствор в холодильнике, он устойчив в течение 3 — 5 дней, при пожелтении для использования не пригоден.
В колбу емкостью 50 мл вносят 2 мл раствора и раствор доводят до метки 15 М серной кислотой.
8. Серная кислота 15 М. К 100 мл воды осторожно при охлаждении приливают 900 мл серной кислоты удельного веса 1,80 — 1,82.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 — 2 л/мин. протягивают через неподвижный слой силикагеля, помещенный в трубку (см. рис. 6). Продолжительность отбора 20 — 30 мин.
При концентрациях формальдегида в воздухе, превышающих допустимые, отбор проб можно проводить в 6 мл воды, помещенных в поглотительные приборы с пористой пластинкой: N 1, со скоростью до 0,5 л/мин.
б) Для определения среднесуточной концентрации воздух протягивают через слой силикагеля со скоростью до 1 л/мин. в течение суток непрерывно. Отбор проб допустимо проводить 6 — 12 раз с перерывами в 2 — 4 часа в течение 30 мин. в один и тот же поглотительный прибор.
Ход анализа
Силикагель переносят в колориметрическую пробирку, добавляют 5 мл дистиллированной воды и нагревают при 50 °C в течение 30 мин. (при закрытой пробке). Периодически перемешивают, добавляют к пробе 2 мл раствора хромотроповой кислоты, пробирки помещают в кипящую водяную баню на 30 мин.
Одновременно готовят стандартную шкалу, при этом наносят стандартный раствор на силикагель (табл. 73).
Таблица 73
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,5 |
0,7 |
1,0 |
2 |
3 |
5 |
|
Дистиллированная вода, мл |
4,5 |
4,3 |
4 |
3 |
2 |
0 |
|
|
Содержание формальдегида, мкг |
0 |
0,5 |
0,7 |
1,0 |
2 |
3 |
5 |
Во все пробирки шкалы добавляют раствор хромотроповой кислоты и повторяют аналогичные операции.
По охлаждении через 30 — 40 мин. сравнивают интенсивность окраски проб с окраской шкалы.
При отборе пробы в жидкую поглотительную среду берут на анализ по 2 мл раствора из каждого поглотительного прибора.
Добавляют по 2 мл хромотроповой кислоты и помещают в кипящую водяную баню на 30 — 40 мин. По охлаждении растворы доливают до 5 мл 9 М раствором серной кислоты, помещают в кюветы с толщиной слоя 1 см и фотометрируют при длине волны 570 нм по сравнению с контролем, который готовят аналогично и одновременно с пробой. Содержание формальдегида определяют по заранее построенному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 74).
Таблица 74
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,8 |
1,6 |
1,4 |
1,2 |
1 |
0 |
|
Содержание формальдегида, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1 |
2 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
Примечания. 1. Очистка хромотроповой кислоты. Хромотроповую кислоту растворяют без нагревания в дистиллированной воде, фильтруют и выпаривают на водяной бане до начала кристаллизации. Охлаждают, отсасывают, промывают небольшим количеством холодной воды, а затем спиртом. Очищенную хромотроповую кислоту сушат в вакуум-эксикаторе и хранят в темной склянке.
2. Раздельное определение формальдегида и метилового спирта см. выше.
ОПРЕДЕЛЕНИЕ ФОРМАЛЬДЕГИДА
С СОЛЯНОКИСЛЫМ ФЕНИЛГИДРАЗИНОМ [3]
Принцип определения
При взаимодействии формальдегида с солянокислым фенилгидразином в присутствии окислителя в щелочной среде образуется соединение, окрашенное в красный цвет. Интенсивность окраски пропорциональна количеству формальдегида.
Чувствительность определения 0,2 мкг в анализируемом объеме раствора.
Аммиак и фенол не мешают определению, если присутствуют в растворе в пятикратном количестве по отношению к формальдегиду.
Реактивы
1. Поглотительный раствор, 50% изопропиловый спирт.
2. Ферроцианид калия, 5% раствор.
3. Солянокислый фенилгидразин, 5% раствор. Навеску соли растворяют в горячей воде и раствор фильтруют.
4. Едкий натр, 10% раствор.
5. Остальные реактивы см. выше.
Отбор пробы
Воздух со скоростью 0,5 — 1 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 6 мл поглотительного раствора.
Ход анализа
В колориметрическую пробирку вносят 5 мл пробы, приливают 0,2 мл 5% раствора солянокислого фенилгидразина, перемешивают и оставляют на 15 мин. Затем вносят по 0,1 мл 5% раствора ферроцианида калия и взбалтывают. Через 10 мин. добавляют 1 мл 10% раствора едкого натра, взбалтывают и через 30 мин. фотометрируют в кювете с толщиной слоя 1 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание формальдегида в анализируемом растворе определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 75).
Таблица 75
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
— |
— |
— |
— |
— |
|
Исходный стандартный раствор, мл |
0 |
— |
— |
— |
— |
— |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
4,98 |
4,96 |
4,94 |
4,92 |
4,9 |
|
Содержание формальдегида, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Все пробирки шкалы обрабатывают аналогично пробам. Измеряют оптическую плотность и строят график. Шкалой стандартов можно пользоваться и для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
Примечание. При раздельном определении формальдегида и метилового спирта строят два калибровочных графика: один по формальдегиду и второй — по метиловому спирту после его окисления.
Пробу делят пополам. В первой половине определяют формальдегид по величине оптической плотности (Д) по графику 1, построенному по формальдегиду. Во второй половине пробы проводят окисление метилового спирта до формальдегида и также измеряют величину оптической плотности (Д1). Разность оптических плотностей (Д1 — Д) соответствует величине оптической плотности (Д2), т.е. интенсивности окраски, полученной за счет окислившегося метилового спирта. В соответствии со значением Д2 по калибровочному графику 2, построенному по метиловому спирту, находят концентрацию метилового спирта. Полученные результаты удваивают.
ФУРФУРОЛ
Бесцветная жидкость, быстро буреющая на воздухе, с запахом миндаля, температура кипения 161,7 °C, плотность при 20 °C 1,59. Фурфурол растворим в воде, спирте, эфире и других растворителях.
Фурфурол является нервным ядом, вызывает судороги и паралич.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ФУРФУРОЛА С АНИЛИНОМ [2]
Принцип определения
При взаимодействии фурфурола с анилином в уксуснокислой среде образуется дианилид оксиглутаконового альдегида, окрашивающий раствор в розовый цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Альдегиды, спирты, ацетон, хлориды, минеральные кислоты не мешают определению.
Реактивы
1. Поглотительный раствор — этанол и вода в отношении 1:1.
2. Стандартный раствор фурфурола. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 10 — 15 мл поглотительного раствора. Колбу взвешивают и в нее вносят 2 — 3 капли фурфурола и снова взвешивают. По разности двух взвешиваний устанавливают количество фурфурола. Объем жидкости в колбе доводят до метки поглотительным раствором, содержимое колбы перемешивают, вычисляют количество фурфурола в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг фурфурола. Раствор доводят до метки поглотительным раствором.
3. Этанол 96°.
4. Уксусная кислота 80 — 100%, раствор.
5. Раствор анилина в уксусной кислоте:
в мерную колбу емкостью 50 мл помещают 5 мл анилина и до метки добавляют уксусную кислоту, раствор тщательно перемешивают.
Отбор пробы
Для определения разовой концентрации протягивают воздух со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащим 5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. при охлаждении.
Ход анализа
В пробирку, в которую вливают 0,5 мл раствора анилина в уксусной кислоте, вносят 4 мл пробы. Содержимое пробирки тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение производят при длине волны 530 нм.
Содержание фурфурола в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 76).
Таблица 76
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
4 |
3,95 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
|
Содержание фурфурола, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения. Шкалу готовят в колориметрических пробирках одновременно с пробой.
Расчет см. выше.
АКРОЛЕИН
|
CH2 =CH·CHO |
Мол. вес 56,06 |
Бесцветная летучая жидкость с неприятным запахом, температура кипения 52,5 °C, температура плавления — 87,7 °C; плотность 0,84. Акролеин растворим в воде, хорошо растворяется в спирте и других органических растворителях. Образуется при неполном сгорании жиров, масел, глицерина, встречается в выхлопных газах, в литейных цехах и т.п. Вызывает резкое раздражение верхних дыхательных путей, глаз.
Предельно допустимые концентрации: максимальная разовая — 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ АКРОЛЕИНА С 4-ГЕКСИЛРЕЗОРЦИНОМ [4, 8]
Принцип определения
При взаимодействии акролеина с 4-гексилрезорцином в присутствии хлорной или уксуснокислой ртути образуется соединение, окрашенное в голубой цвет.
Чувствительность определения 1 мкг акролеина в анализируемом объеме раствора.
Определению не мешают двуокись азота в количествах до 5 мкг, кротоновый альдегид до 2 мкг. Не мешают определению сернистый газ, ароматические углеводороды, фенолы, эфиры, кетоны.
Реактивы
1. Поглотительный раствор. Готовят смешением растворов 4-гексилрезорцина, хлорида или ацетата ртути, трихлоруксусной кислоты и этилового спирта в отношениях 1:2:50:50.
Поглотительный раствор устойчив в течение дня.
2. Исходный стандартный раствор готовят в этаноле с содержанием 100 мкг/мл. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разведением исходного.
3. 4-Гексилрезорцин, 50% раствор в этаноле.
4. Хлорная ртуть, 3% раствор в этаноле.
5. Ацетат ртути (двузамещенная соль), 3% раствор в этаноле.
6. Трихлоруксусная кислота, насыщенный раствор. 100 г кислоты (х. ч.) растворяют в 10 мл дистиллированной воды при нагревании на водяной бане, затем объем доводят до 70 мл водой. Раствор употребляют свежеприготовленный.
7. Этиловый спирт, ректификат.
Отбор пробы
Для определения максимальной разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 5 мл поглотительного раствора. Продолжительность отбора — 60 мин.
Ход анализа
Определение акролеина проводят в день отбора пробы или на следующий день. Пробу хранят в холодильнике. При анализе температура раствора должна быть 18 — 20 °C. Содержимое каждого поглотительного прибора доводят до первоначального объема этиловым спиртом, переносят в пробирки, нагревают в течение 30 мин. на водяной бане при 60 °C. По охлаждении через 15 мин. фотометрируют окрашенный раствор в кюветах с толщиной слоя 1 см при длине волны 605 нм по сравнению с контролем. Содержание акролеина в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 77).
Таблица 77
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Этиловый спирт, мл |
1,8 |
1,7 |
1,6 |
1,4 |
1,2 |
1,0 |
0,8 |
|
Содержание акролеина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
При приготовлении шкалы в каждую пробирку вносят 0,5 мл раствора 4-гексилрезорцина, 0,2 мл раствора хлорной ртути или 0,2 мл раствора ацетата ртути, 2,5 мл раствора трихлоруксусной кислоты.
После тщательного перемешивания пробирки помещают на 30 мин. в водяную баню, нагретую до 60 °C, фотометрируют и строят калибровочный график.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ АКРОЛЕИНА ФЛУОРЕСЦЕНТНЫМ МЕТОДОМ [5]
Принцип определения
При взаимодействии акролеина с м-фенилендиамином образуется производное, которое выделяют из смеси в тонком слое силикагеля. Полученное соединение в ультрафиолете флуоресцирует голубым светом.
Чувствительность определения — 0,1 мкг акролеина в 5 мл элюата. Определению не мешают: предельные, непредельные, ароматические альдегиды. Кротоновый и уксусный альдегиды также дают флуоресцирующие соединения, но зоны локализаций этих соединений расположены на других уровнях на хроматограмме и определению акролеина не мешают.
Аппаратура
1. Флуорометр, лампа УФ-ПР К-4 или ПР К-2 со стеклом Вуда.
Реактивы
1. Поглотительный раствор. Гидроксиламин солянокислый, 0,25% раствор.
2. Исходный стандартный раствор акролеина готовят с содержанием 1 — 2 мг/мл в воде. Рабочий стандартный раствор готовят разбавлением исходного 0,25% раствором гидроксиламина. Рабочий стандартный раствор содержит 2 мкг/мл акролеина.
Исходный и рабочий растворы готовят перед употреблением.
3. М-Фенилендиамин, 0,5% раствор, приготовленный в 1 н. растворе соляной кислоты. Хранят в темном месте. Срок хранения 3 — 4 дня.
4. Соляная кислота, 5 н., 1 н. растворы.
5. Аммиак, 25% раствор.
6. Хлороформ.
7. Изопропиловый спирт.
8. Уксусная кислота, ледяная.
9. Гидроксиламин солянокислый, 0,25% раствор.
10. Спирт этиловый, ректификат, осушенный.
11. Системы растворителей: диметилформамид — бензол 1:2 или четыреххлористый углерод — этиловый спирт 1:5 или хлороформ — этиловый спирт 1:4 или этиловый спирт — хлороформ 4:1 по объему.
12. Силикагель КСМ, КСК, очищенный. Силикагель заливают соляной кислотой 1:1 и выдерживают в течение 18 — 20 ч, промывают водой. Добавляют азотной кислоты 1:1 и кипятят в течение 2 — 3 ч. Далее промывают проточной водопроводной водой, затем дистиллированной до нейтральной реакции промывных вод. Высушивают при 130 °C в течение 4 — 6 ч.
13. Кальций сернокислый (гипс), х.ч. или специально приготовленный. Для этого водный раствор хлористого кальция смешивают с эквимолярным количеством серной кислоты при 70 — 80 °C. Сульфат кальция отделяют, промывают водой до нейтральной реакции и высушивают при 120 °C.
14. Хроматографические пластинки «Silufol» или специально приготовленные. Для этого берут стеклянную пластинку размером 9 x 12 см, промывают хромовой смесью, дистиллированной водой и высушивают в вертикальном положении. Перед нанесением сорбционной массы пластину протирают этиловым спиртом. Берут 5 г силикагеля с зернами величиной 0,1 — 0,07 мм, смешивают с 0,5 г гипса и добавляют 10 мл воды. К полученной смеси добавляют еще 8 мл воды и тщательно перемешивают. 10 г сорбционной массы наносят на пластину и равномерно распределяют по поверхности. Высушивают в течение часа при 100 °C. Хранят над хлористым кальцием. Хроматографические пластины, обработанные люминесцирующим реактивом, употреблять не рекомендуется.
Отбор пробы
а) Для определения максимальной разовой концентрации акролеина воздух со скоростью 0,5 л/мин. протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 5 мл 0,25% раствора солянокислого гидроксиламина. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают через два последовательно соединенных поглотительных прибора Зайцева, наполненных по 10 мл 0,25% раствора солянокислого гидроксиламина, со скоростью 0,2 — 0,3 л/мин. 6 — 12 раз в одну и ту же поглотительную систему с перерывами в 2 — 4 ч.
Ход анализа
Жидкость из каждого поглотительного прибора переносят в пробирки. Добавляют по 1 мл 0,5% раствора м-фенилендиамина и по 1 мл 5 н. раствора соляной кислоты. Содержимое пробирок перемешивают и помещают в кипящую водяную баню на 15 мин. По охлаждении содержимое пробирок переносят в делительные воронки, вносят по 0,5 мл 25% раствора аммиака и по 5 мл хлороформа.
Смесь перемешивают в течение 1 — 2 мин. и нижний слой хлороформа фильтруют через бумажные фильтры в сухие пробирки или выпарительные чашки. Хлороформ удаляют на водяной бане при температуре не выше 60 °C. Сухие остатки обрабатывают 0,1 мл хлороформа и по 0,05 мл с помощью микропипетки наносят на стартовую линию на хроматографическую пластину. Предварительно на пластине намечают точки на расстоянии 1 см от края и на расстоянии 2,5 см друг от друга. После высыхания (удаления хлороформа) пластину помещают в хроматографическую камеру, в которую заранее вносят одну из систем растворителей. Смесь диметилформамида с бензолом или хлороформа с этиловым спиртом помещают в делительную воронку, перемешивают и вносят в хроматографическую камеру с тем расчетом, чтобы пластина погружалась в систему не более чем на 0,5 см. После того как линия фронта будет на расстоянии 1 см от верхнего края пластины, хроматограмму вынимают из камеры, высушивают на воздухе и просматривают в ультрафиолетовом свете (с фильтром N 3). Флуоресцирующие голубым светом зоны локализации, соответствующие акролеину, определяют по свидетелю, нанесенному на пластину одновременно с пробой (так как в разных системах растворителей и на разных пластинах Rf производных акролеина различна).
Участки силикагеля с флуоресцирующими производными акролеина снимают и помещают в пробирки. В пробирки вносят по 6 мл 1 н. раствора соляной кислоты, перемешивают в течение 1 — 2 мин. и центрифугируют. Интенсивность флуоресценции элюатов измеряют на флуориметре с желтым светофильтром. Содержание акролеина находят по калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов в соответствии с табл. 78.
Таблица 78
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,5 |
0,6 |
0,7 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,5 |
4,4 |
4,3 |
|
Содержание акролеина, мкг |
0 |
0,1 |
0,2 |
0,4 |
0,8 |
1,0 |
1,2 |
1,4 |
Полученные таким образом растворы 1 — 2…7 обрабатывают аналогично пробе. Для построения калибровочного графика сухие остатки растворяют в 0,1 мл хлороформа и в количестве по 0,05 мл наносят на пластины, что соответствует 0,05 — 0,1 — 0,2 — 0,4 — 0,5 — 0,6 и 0,7 мкг акролеина. Раствор 1 н. соляной кислоты устанавливают на 6% по шкале флуориметра; этот раствор служит контролем. Затем измеряют интенсивность флуоресценции всех элюатов, показание контроля вычитают. График строят на основании средних данных из 5 — 6 шкал.
Расчет анализа см. выше.
АЦЕТОН
Бесцветная жидкость с характерным запахом. Температура кипения 56,24 °C, плотность при 20 °C 0,791. Хорошо растворим в воде, спирте, эфире и других органических растворителях. Ацетон является хорошим растворителем жира, резины, воска, нитроцеллюлозы. Смесь паров ацетона с воздухом обладает взрывчатыми свойствами.
Ацетон действует как наркотик, поражает центральную нервную систему.
Предельно допустимая концентрация: максимальная разовая и среднесуточная 0,35 мг/м3.
ОПРЕДЕЛЕНИЕ АЦЕТОНА С САЛИЦИЛОВЫМ АЛЬДЕГИДОМ [11]
Принцип определения
При взаимодействии ацетона с салициловым альдегидом в щелочной среде образуется соединение, окрашивающее раствор в желтый цвет, по интенсивности которого определяют содержание ацетона.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Спирты, альдегиды до концентрации 7 мг/л не мешают определению.
Реактивы
1. Силикагель КСК, величина зерен 0,5 — 2 мм. Силикагель обрабатывают соляной кислотой, разбавленной водой в отношении 1:1 при нагревании в течение 3 — 4 ч, затем промывают водой, высушивают при 105 °C и активируют при 200 °C в течение 30 мин. Силикагель проверяют на отсутствие ацетона. Бывший в употреблении силикагель регенерируют, промывают и активируют.
2. Поглотительный раствор — дистиллированная вода.
3. Стандартный раствор ацетона: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую влито 15 — 20 мл воды. Колбу взвешивают, вносят в нее 2 — 3 капли свежеперегнанного ацетона и снова взвешивают. По разности двух взвешиваний устанавливают количество ацетона в колбе. Раствор встряхивают, доводя до метки водой. Высчитывают количество ацетона в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят в мерной колбе емкостью 100 мл, в нее вносят такое количество исходного раствора, которое содержит 1000 мкг/мл ацетона. Раствор доводят до метки водой.
Применяют только свежеприготовленный раствор.
4. Салициловый альдегид 2,18 мл свежеперегнанного альдегида (температура кипения 193 °C), вносят в мерную колбу емкостью 50 мл, в которую влито 10 — 20 мл этанола, и добавляют до метки этанолом.
5. Этанол 96°.
6. Едкое кали, 50% раствор.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух через два поглотительных прибора Института имени Ф.Ф. Эрисмана, к которым присоединяют прибор для улавливания переброшенных частиц. Каждый прибор содержит по 3 мл силикагеля, скорость отбора через «кипящий слой» силикагеля — 5 л/мин.
При отборе пробы в жидкую поглотительную среду воздух протягивают со скоростью 0,3 л/мин., через поглотительный прибор с пористым фильтром N 1, содержащий 4 мл воды. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух протягивают в кипящий слой силикагеля со скоростью 1 — 2 л/мин. непрерывно в течение суток. Допустимо отбор пробы проводить 6 — 12 раз с перерывами в 2 — 4 ч по 30 мин. в один и тот же поглотительный прибор.
Ход анализа
Силикагель пересыпают в пробирки с притертыми пробками из каждого прибора отдельно. Переброшенные зерна из уловителя пересыпают в пробирки второго прибора.
Силикагель в каждой пробирке заливают 6 мл воды и помещают на водяную баню, нагретую до 40 °C, на 30 мин., производя встряхивание раствора через 5 — 10 мин.
По охлаждении раствора 2 мл пробы переводят в пробирку и наливают 1 мл 50% раствора едкого кали и 0,5 мл салицилового альдегида. Содержимое пробирки встряхивают и помещают на 25 мин. в водяную баню, нагретую до 70°.
После охлаждения раствора проводят определение ацетона, сравнивая интенсивность окраски пробы со шкалой стандартов (табл. 79).
Таблица 79
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода, мл |
2 |
1,9 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание ацетона, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе одновременно с ней.
При отборе проб в воду отбирают 2 мл пробы и обрабатывают аналогично, используя ту же шкалу стандартов.
Расчет см. выше.
ЦИКЛОГЕКСАНОН
Бесцветная маслянистая жидкость с запахом мяты, температура кипения 155,4 °C, плотность пара по отношению к воздуху 3,4. Почти не растворима в воде, хорошо растворима в ряде органических растворителей.
Циклогексанон раздражающе действует на слизистые оболочки глаз, горла и носа.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ ЦИКЛОГЕКСАНОНА С ФУРФУРОЛОМ [7]
Принцип определения
При взаимодействии циклогексанона и фурфурола в щелочной среде образуется фурфурилиденциклогексанон. Последний, вступая в соединение с серной кислотой, окрашивает раствор в малиновый цвет, по интенсивности которого определяют содержание циклогексанона.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Другие кетоны мешают определению.
Реактивы
1. Поглотительный раствор — этанол, разбавленный водой в отношении 1:1.
2. Стандартный раствор циклогексанона: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 5 — 10 мл поглотительного раствора, и колбу взвешивают. Затем в колбу вносят 0,1 — 0,2 мл циклогексанона и вновь взвешивают. Содержимое колбы доводят до метки поглотительным раствором и взбалтывают. По разности двух взвешиваний устанавливают количество циклогексанона в колбе и высчитывают его содержание в 1 мл.
Рабочий стандартный раствор с содержанием 10 мкг/мл циклогексанона готовят в мерной колбе емкостью 50 мл. В нее вносят такое количество исходного раствора, в котором содержится 500 мкг циклогексанона. Объем жидкости в колбе доводят до метки поглотительным раствором.
3. Этанол, 96°.
4. Фурфурол свежеперегнанный, из него готовят 0,3% водный раствор; он устойчив в течение 2 — 3 недель.
5. Натр едкий, 25% раствор.
6. Серная кислота, уд. вес 1,84.
Отбор пробы
Для определения разовой концентрации воздух протягивают со скоростью 0,5 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 3 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин. При отборе поглотительный прибор помещают в баню со льдом.
Ход анализа
В пробирку помещают 2 мл пробы, добавляют 0,2 мл 0,3% раствора фурфурола и 0,2 мл 25% раствора едкого натра. Раствор в пробирке встряхивают и оставляют в покое в течение часа. По истечении этого времени в пробирку добавляют 3 мл серной кислоты уд. веса 1,84. Содержимое пробирки встряхивают и по охлаждении измеряют оптическую плотность в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Определение проводят при длине волны 555 нм. Содержание циклогексанона в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов.
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробой (табл. 80).
Расчет см. выше.
Таблица 80
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,07 |
0,09 |
0,1 |
0,15 |
0,2 |
0,3 |
|
Поглотительный раствор, мл |
2 |
1,95 |
1,93 |
1,91 |
1,9 |
1,85 |
1,8 |
1,7 |
|
Содержание циклогексанона, мкг |
0 |
0,5 |
0,7 |
0,9 |
1,0 |
1,5 |
2,0 |
3,0 |
АЦЕТОФЕНОН
(Метилфенилкетон, ацетилбензол)
Жидкость без цвета, с резко выраженным запахом. Температура плавления 19,7 °C; температура кипения 202,3 °C. Не растворим в воде, хорошо растворим в этаноле, эфире и других органических растворителях. Ацетофенон токсичен.
Предельно допустимые концентрации: максимальная разовая и среднесуточная — 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ АЦЕТОФЕНОНА С М-ДИНИТРОБЕНЗОЛОМ [10]
Принцип определения
При взаимодействии ацетофенона с М-динитробензолом в щелочной среде образуется продукт, окрашенный в розовый цвет.
Чувствительность определения — 2 мкг в анализируемом объеме раствора.
Ацетон мешает определению в количествах, превышающих 100 мкг. Бензол, изопропилбензол, гидроперекись изопропилбензола, ![]() -метилстирол, стирол, фенол не мешают определению.
-метилстирол, стирол, фенол не мешают определению.
Реактивы
1. Силикагель КСК, МСК, КСМ, МСМ с зернами величиной 0,25 — 2 мм, обработанных, как указано выше. Каждую партию силикагеля проверяют на отсутствие ацетофенона.
2. Стандартный раствор ацетофенона. Исходный стандартный раствор готовят путем растворения точной навески ацетофенона в этаноле. Рабочий стандартный раствор с содержанием 10 мкг ацетофенона в 1 мл готовят соответствующим разбавлением исходного, перед употреблением.
3. М-динитробензол, 1% раствор в этаноле готовят в малом объеме (10 мл), раствор устойчив в течение суток.
А. Калий едкий, 30% раствор.
Отбор пробы
А) Воздух со скоростью до 10 л/мин. протягивают в «кипящий» слой силикагеля. Для этого по 3 мл силикагеля помещают в два последовательно соединенных поглотительных прибора Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана, соединенных с поглотителем для задержания переброшенных частиц. Отбор проб проводят при охлаждении. Отбирают не менее часа.
Б) Для определения среднесуточной концентрации воздух со скоростью 5 — 10 л/мин. протягивают в кипящий слой силикагеля непрерывно в течение суток.
Допустимо проводить отбор 6 — 12 раз с перерывами в 4 или 2 ч.
При отборе поглотительный прибор помещают в охладительную смесь.
Ход анализа
Содержимое поглотительных приборов переносят в отдельные пробирки с притертыми пробками. Во вторую пробирку помещают переброшенные частицы силикагеля.
Заливают силикагель по 4 мл этанола. Периодически встряхивают в течение 30 мин. Добавляют по 0,2 мл раствора М-динитробензола, по 0,3 мл раствора едкого кали и по 0,5 мл воды.
Содержимое пробирок перемешивают. Одновременно готовят шкалу стандартов. Для этого в серию пробирок вносят по 3 мл силикагеля, который употреблен при анализе, наносят на силикагель рабочий стандартный раствор, перемешивают. Одновременно готовят контроль (табл. 81).
Таблица 81
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
2,0 |
2,5 |
|
Этанол, мл |
По 4 мл |
|||||||||
|
Содержание ацетофенона, мкг |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
15 |
20 |
25 |
В каждую пробирку шкалы и в контроль вносят по 0,2 мл раствора М-динитробензола и другие реактивы аналогично пробе.
Через час сравнивают интенсивность окраски в пробах с окраской стандартной шкалы в растворах над силикагелем.
Расчет см. выше.
ДИКЕТЕН
|
|
Мол. вес 84,0 |
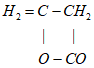
Бесцветная жидкость с острым запахом, растворимая в органических растворителях, не растворимая в воде. Температура кипения 127,4 °C, плотность 1,089. Дикетен весьма реакционноспособен, легко подвергается полимеризации.
Пары дикетена вызывают резкое раздражение глаз и дыхательных путей. Характерными являются бронхиты с пневматическими очагами, отеки легких. Предельно допустимые концентрации: максимальная разовая — 0,007 мг/м3.
ОПРЕДЕЛЕНИЕ ДИКЕТЕНА С РЕЗОРЦИНОМ [9]
Принцип определения
Продукт реакции дикетена с серной кислотой, взаимодействуя с резорцином, дает сине-фиолетовую люминесценцию при облучении ультрафиолетовым излучением.
Чувствительность определения — 0,02 мкг в анализируемом объеме раствора.
Определению мешает кетен.
Ацетон до 170 мкг, уксусный ангидрид до 70 мкг, уксусная кислота, метан, этилен, окись и двуокись углерода не мешают определению.
Реактивы
1. Поглотительный раствор: серная кислота, приготовленная в отношении 8:2. Для приготовления 100 мл раствора в колбу вводят 20 мл дистиллированной воды, а затем добавляют 80 мл концентрированной серной кислоты. Полученный раствор охлаждают и проверяют на отсутствие люминесценции.
2. Дикетен перегнанный.
Перегонку дикетена производят под вакуумом при остаточном давлении 50 мм рт. ст. Стабилизируют дикетен сернокислым цинком из расчета 1 г на 100 мл. Хранить дикетен следует в холодильнике.
3. Стандартные растворы дикетена. Для приготовления исходного раствора дикетена в мерную колбу емкостью 25 — 50 мл наливают 5 — 10 мл дистиллированной воды и взвешивают на аналитических весах.
В колбу вносят 1 — 2 капли дикетена и снова взвешивают. Объем в колбе доводят до метки водой и хорошо перемешивают. По разности в весе определяют содержание дикетена в 1 мл раствора. Из исходного раствора готовят стандартный раствор с содержанием 2 мкг/мл, а из него рабочий стандартный раствор с содержанием 0,2 мкг/мл. Рабочий стандартный раствор готовят на серной кислоте, приготовленной в отношении 8:2.
Стандартный раствор устойчив в течение 5 суток при хранении в холодильнике.
3. Серная кислота, уд. вес 1,84.
4. Резорцин. Если резорцин загрязнен или окрашен, его очищают возгонкой. Для этого 2 — 3 г резорцина помещают на часовое стекло, которое покрывают другим таким же стеклом. Между стеклами прокладывают фильтровальную бумагу с большим количеством отверстий. Возгонку резорцина проводят на песчаной бане при 115 — 120 °C. При возгонке верхнее стекло охлаждают фильтровальной бумагой, смоченной водой. Образующиеся между фильтровальной бумагой и верхним стеклом прозрачные кристаллы резорцина собирают и хранят в склянке из темного стекла с притертой пробкой.
5. 1% водный раствор резорцина. Раствор хранят в темной склянке с притертой пробкой, он устойчив в течение 1 — 2 мес.
6. Сульфат цинка, прокаленный для удаления влаги.
7. Дистиллированная вода, нелюминесцирующая под ультрафиолетовым светом.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 л/мин. протягивают через поглотительный прибор Полежаева, содержащий 1 мл поглотительного раствора, в течение 20 — 30 мин.
Ход анализа
Готовят шкалу стандартов в пробирках из нелюминесцирующего стекла (табл. 82).
Таблица 82
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
Рабочий стендартный раствор, мл |
0 |
0,1 |
0,2 |
0,3 |
0,4 |
0,5 |
0,6 |
0,7 |
0,8 |
0,9 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,9 |
0,8 |
0,7 |
0,6 |
0,5 |
0,4 |
0,3 |
0,2 |
0,1 |
0 |
|
Содержание дикетена, мкг |
0 |
0,02 |
0,04 |
0,06 |
0,08 |
0,1 |
0,12 |
0,14 |
0,16 |
0,18 |
0,20 |
Одновременно в поглотители с пробами и в пробирки шкалы добавляют по 0,1 мл 1% раствора резорцина и хорошо перемешивают. Содержимое поглотителей переливают в пробирки из нелюминесцирующего стекла и через 15 мин. сопоставляют интенсивность свечения пробы со шкалой стандартов на установке для люминесцентного визуального анализа (лампа ПРК-4, светофильтр УФС-3). Шкалой стандартов можно пользоваться в течение 4 — 5 ч. Контроль не должен давать люминесценции. При наличии в нем люминесценции следует проверить чистоту пробирок и реактивов.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение формальдегида. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
2. Алексеева М.В. Определение фурфурола. В кн.: Определение атмосферных загрязнений. М., «Медгиз», 1963.
3. Бензина Г.И. Фотометрический метод определения формальдегида в воздухе. Гиг. и сан., 1968, 6.
4. Ипатова С.А. К вопросу определения акролеина с 4-гексилрезорцином. Гиг. и сан., 1971, 1.
5. Ипатова С.А., Деянова Е.В. Определение акролеина в воздухе флюоресцентным методом. Гиг. и саи., 1973, 10.
6. Качмар Е.Г., Хрусталева В.А. Применение зерненных сорбентов для отбора проб атмосферного воздуха. Гиг. и сан., 1969, 9.
7. Масленников А.С. Определение циклогексанона в воздухе. Заводская лаборатория, 1954, 1.
8. Новый спектрофотометрический метод определения акролеина в дымовых газах и атмосфере. Реферат (пер. с англ. Л.И. Вагранской). Гиг. и сан., 1963, 9.
9. Определение дикетена в воздухе. Технические условия на методы определения вредных веществ в воздухе. В. 5. М., «Медицина», 1968.
10. Хрусталева В.А. Определение ацетофенона в воздухе. Гиг. и сан., 1961, 12.
11. Шихваргер Ф.Д. Количественное определение ацетона в атмосферном воздухе по реакции с салициловым альдегидом. Заводская лаборатория, 1951, т. 17, с. 11.
Глава XII
АНГИДРИДЫ, ОРГАНИЧЕСКИЕ КИСЛОТЫ
МАЛЕИНОВЫЙ АНГИДРИД
|
|
Мол. вес 98,06 |
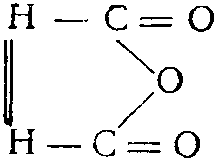
Бесцветное кристаллическое вещество с температурой плавления 52 — 53 °C, температурой кипения 202 °C, обладает резким запахом, растворим в воде, спирте, ацетоне. Применяется при получении полиэфирных смол, при диеновых синтезах. Обладает местным раздражающим действием, вызывает конъюнктивит, раздражение верхних дыхательных путей, кожи.
Предельно допустимые концентрации: максимальная разовая — 0,2 мг/м3, среднесуточная — 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ПАРОВ МАЛЕИНОВОГО АНГИДРИДА ОКИСЛЕНИЕМ [3]
Принцип определения
Метод основан на окислении малеиновой кислоты, образующейся в водной среде из малеинового ангидрида, до винной кислоты. Окисление проходит по месту двойной связи, при этом фиолетовый цвет раскисляющегося перманганата переходит в желтовато-бурый.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора. Определению мешают непредельные соединения: фталиевый ангидрид, эпихлоргидрин; гликоли не мешают определению.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор малеинового ангидрида с содержанием 100 мкг/мл. Из него разбавлением в 10 раз готовят рабочий стандартный раствор с содержанием 10 мкг/мл.
3. Перманганат калия, 0,04% раствор.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,2 — 0,3 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 3 мл дистиллированной воды.
Продолжительность отбора 20 — 30 мин.
Ход анализа
Из каждого поглотительного прибора 2 мл пробы берут на анализ. Одновременно готовят шкалу стандартов (табл. 83).
Таблица 83
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Вода дистиллированная, мл |
2 |
1,95 |
1,90 |
1,8 |
1,6 |
1,4 |
1,2 |
1,0 |
|
Содержание малеинового ангидрида, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
Во все пробирки шкалы и пробы добавляют по 0,1 мл 0,04% раствора марганцевокислого калия. Через 5 — 10 мин. проводят сравнение окраски пробы со шкалой.
Расчет см. выше.
Примечание. При наличии в воздухе аэрозоля малеинового ангидрида отбор проб проводят на фильтр АФА.
ОДНООСНОВНЫЕ КАРБОНОВЫЕ КИСЛОТЫ C1 — C9
CнH2нO2
Прозрачные бесцветные жидкости. Кислоты C1 — C3 обладают резким запахом, C — C — неприятным прогорклым запахом. Кислоты C4 — C9 хорошо растворимы в воде, но с увеличением молекулярного веса растворимость в воде уменьшается. Все кислоты хорошо растворимы в этаноле и эфире.
Токсические свойства кислот по силе и характеру воздействия неодинаковы. Муравьиная и уксусная кислоты сильно раздражают верхние дыхательные пути; эти кислоты, попадая на кожу, вызывают ожоги, пропионовая кислота вызывает умеренное раздражение кожи. Раздражающее действие среднемолекулярных кислот C1 — C9 выражено слабо.
Попадая в организм, кислоты вызывают изменение внутренних органов, крови, сосудисто-сердечной и нервной системы и др. Некоторые свойства карбоновых кислот представлены в табл. 84.
Таблица 84
НЕКОТОРЫЕ ФИЗИЧЕСКИЕ СВОЙСТВА И ПРЕДЕЛЬНО ДОПУСТИМЫЕ
КОНЦЕНТРАЦИИ КАРБОНОВЫХ КИСЛОТ
|
Название |
Формула |
Молекулярный вес |
Удельный вес (20°) |
Температура кипения |
ПДК мг/м3 |
|
|
максимальная разовая |
среднесуточная |
|||||
|
Муравьиная |
C1H2O2 |
46,03 |
1,2200 |
100, 7 |
||
|
Уксусная |
C2H4O2 |
60,05 |
1,0490 |
118,1 |
0,2 |
0,06 |
|
Пропионовая |
C3H6O2 |
74,08 |
0,9934 |
141,1 |
||
|
Масляная |
C4H8O2 |
88, 10 |
0,9587 |
163,5 |
0,015 |
0,01 |
|
Валериановая |
C5H10O2 |
102,14 |
0,9420 |
186,3 |
0,03 |
0,01 |
|
Капроновая |
C6H12O2 |
116,16 |
0,9290 |
205,0 |
0,01 |
0,005 |
|
Энантовая |
C7H14O2 |
130,19 |
0,9184 |
2283,0 |
||
|
Каприловая |
C8H16O2 |
144,22 |
0,9100 |
237,5 |
||
|
Пеларгоновая |
C9H18O2 |
158,24 |
0,9055 |
254,0 |
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА ОДНООСНОВНЫХ
КАРБОНОВЫХ КИСЛОТ C1 — C9 [2]
Принцип определения
При взаимодействии кислот с метанолом в присутствии серной кислоты образуются эфиры, которые, вступая в реакцию с гидроксиламином в щелочной среде, превращаются в гидроксаматы. Последние в кислой среде, реагируя с хлорным железом, переходят в железогидроксамовый комплекс, окрашивая раствор в буро-розовый цвет.
Чувствительность фотометрического определения — 10 — 20 мкг (C1 — C3) и (C4 — C9) соответственно в анализируемом объеме раствора.
Чувствительность визуального определения — 2 мкг в анализируемом объеме раствора.
Минеральные кислоты, ацетон, динил, спирты, сложные летучие эфиры жирных кислот определению не мешают.
Реактивы
1. Силикагель СМ для отбора проб, величина зерен от 0,5 до 2 мм. Силикагель предварительно обрабатывают соляной кислотой, разбавленной водой в отношении 1:1 при нагревании до 60 — 80°, периодически помешивая силикагель. Пожелтевшую кислоту несколько раз заменяют свежим раствором. Обработку заканчивают, когда раствор соляной кислоты остается бесцветным. Затем силикагель отмывают от кислоты водой, сушат при 105 °C и активируют при 200 — 250 °C в течение 20 мин. Обработанный силикагель регенерируют описанным выше методом.
2. Стандартный раствор кислот. Готовят исходный стандартный раствор из каждой кислоты в мерных колбах емкостью 25 мл. В колбы вливают 10 — 5 мл этанола, колбы взвешивают, вносят в каждую по 5 — 10 капель кислоты и снова взвешивают, объем раствора доводят до метки этанолом. По разности двух взвешиваний устанавливают в каждой колбе содержание кислоты и высчитывают количество кислоты в 1 мл раствора. Рабочий стандартный раствор должен содержать все исследуемые кислоты по 100 мкг/мл каждой кислоты. Для этого в мерную колбу емкостью 50 мл вносят такое количество исходного раствора каждой кислоты, в котором содержится 5 мг. Объем колбы доводят до метки этанолом.
3. Серная кислота, уд. вес 1,84%, 50% в метаноле.
4. Гидроксиламин солянокислый, 4 н. раствор.
5. Едкий натр, 4 и 0,05 н. раствор свежеприготовленный.
6. Соляная кислота, 0,5 и 4 н. раствор.
7. Хлорное железо окисное 1,6% раствор в 0,5 н. растворе соляной кислоты.
8. Этанол, обработанный щелочью и перегнанный.
9. Метанол осушенный. Обезвоживание метанола проводят металлическим магнием.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух протягивают со скоростью 5 — 6 л/мин. через два последовательно соединенных поглотительных прибора — Яворовской и НИИ гигиены имени Ф.Ф. Эрисмана, наполненных 2 г силикагеля, в «кипящий» слой. Продолжительность отбора 40 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 3 л/мин. протягивают через силикагель в течение суток непрерывно. Допустимо отбор проводить 6 — 12 раз с перерывами в 4 — 2 ч по 20 — 30 мин.
Ход анализа
Силикагель переносят в пробирки из каждого поглотительного прибора отдельно. Переброшенные зерна силикагеля присоединяют к силикагелю из контрольного поглотительного прибора.
Пробу, помещенную в пробирку, заливают 5 мл этанола, встряхивают и оставляют на 60 мин., периодически встряхивая. По истечении этого времени для анализа берут 3 мл и помещают в фарфоровый тигель. Затем кислоту переводят в соли путем титрования пробы 0,05 н. раствором едкого натра в присутствии одной капли фенолфталеина.
Тигель помещают в кипящую водяную баню для удаления этанола. Вместе с этанолом испаряются эфиры, если они присутствуют в пробе.
Сухой остаток подсушивают в эксикаторе или в сушильном шкафу при 80° (эта операция обязательна).
Осадок в тигле постепенно растворяют в 1,5 мл метанола в три приема по 0,5 мл каждый раз, переводя пробу в колориметрическую пробирку. В пробу вносят одну каплю серной кислоты уд. веса 1,84. Содержимое пробирки встряхивают и оставляют на 30 мин., периодически встряхивая жидкость.
Через 30 мин. в пробирку вливают 0,2 мл 4 н. раствора солянокислого гидроксиламина и 0,6 мл 4 н. раствора едкого натра. Раствор встряхивают и оставляют на 15 мин. По истечении указанного времени раствор нейтрализуют 4 н. соляной кислотой по фенолфталеину. Затем вносят 2,5 мл 1,6% раствора хлорида железа окисного. Раствор встряхивают, через 20 — 30 мин. измеряют оптическую плотность раствора в кювете с толщиной слоя 1 см при длине волны 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание кислоты в анализируемом объеме раствора определяют по калибровочному графику, который строят по шкале стандартов (табл. 85).
Таблица 85
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,5 |
|
Метанол, мл |
1,5 |
1,4 |
1,3 |
1,1 |
0,9 |
0,7 |
0,5 |
0,3 |
0 |
|
Содержание кислот, мкг |
0 |
10 |
20 |
40 |
60 |
80 |
100 |
120 |
150 |
Все пробирки шкалы обрабатывают аналогично пробе.
При визуальном определении проводят те же операции, как описано выше, но сухой остаток после подсушивания в сушильном шкафу растворяют в 0,75 мл метанола.
В пробу вносят одну каплю 50% раствора серной кислоты в метаноле и оставляют пробу на 30 мин. для полного образования эфира. Временами раствор встряхивают. Через 30 мин. в тигель вливают 0,1 мл 4 н. раствора гидроксиламина и 0,4 мл 4 н. раствора едкого натра. Раствор перемешивают и через 15 мин. переливают в колориметрическую пробирку. В тигель вливают 1,5 мл 1,6% раствора хлорного железа и его ополаскивают этим раствором. Затем раствор переливают из тигля в пробирку с пробой. Проба окрашивается в буро-розовый цвет.
Через 10 — 20 мин. интенсивность окраски пробы сравнивают со шкалой стандартов, которую готовят одновременно с пробой (табл. 86).
Таблица 86
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,02 |
0,05 |
0,10 |
0,20 |
0,30 |
0,40 |
0,50 |
|
Метанол, мл |
0,75 |
0,73 |
0,70 |
0,65 |
0,55 |
0,45 |
0,35 |
0,25 |
|
Содержание кислот, мкг |
0 |
2 |
5 |
10 |
20 |
30 |
40 |
50 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
РАЗДЕЛЬНОЕ ОПРЕДЕЛЕНИЕ КАРБОНОВЫХ КИСЛОТ
(C1 — C9) ПРИ ПОМОЩИ БУМАЖНОЙ ХРОМАТОГРАФИИ [1]
Принцип определения
Метод основан на переведении кислот в натриевые соли, затем в соответствующие гидроксамовые кислоты, которые разделяют на бумаге нисходящим методом в двух системах растворителей — бутанол, уксусная кислота, вода в соотношении 4:1:5 (для C1 — C4) и бензол, муравьиная кислота, вода в соотношении 1:1:1 (для C5 — C9).
После проявления хроматограммы производные кислот обнаруживаются в виде фиолетовых пятен.
Количественное определение проводят визуально путем сравнения интенсивности окраски пятен проб с окраской «свидетелей», а также фотометрически, после окисления элюированных гидроксамовых кислот до азотистой кислоты и определения последней с сулфаниловой кислотой и ![]() -нафтиламином. Минимальные количества, открываемые на хроматограммах, соответствуют 3 мкг для каждой из кислот C1 — C4 и 5 мкг для каждой из кислот C5 — C9. Определению не мешают минеральные кислоты, спирты, сложные эфиры жирных кислот групп C1 — C9, формальдегид, кетоны, амины C7 — C9, динил.
-нафтиламином. Минимальные количества, открываемые на хроматограммах, соответствуют 3 мкг для каждой из кислот C1 — C4 и 5 мкг для каждой из кислот C5 — C9. Определению не мешают минеральные кислоты, спирты, сложные эфиры жирных кислот групп C1 — C9, формальдегид, кетоны, амины C7 — C9, динил.
Аппаратура
1. Хроматографическая камера, представляющая собой стеклянный цилиндр высотой 50 — 55 см, диаметром 20 — 30 см с хорошо пришлифованной крышкой (см. рис. 26).
2. Стеклянная лодочка.
3. Теплоэлектровентилятор.
4. Стеклянный распылитель.
5. Пробирки высотой 60 мм, диаметр 8 мм с делениями на 0,3 и 0,5 мл.
6. Микропипетки емкостью 0,1 и 0,2 мл с делениями 0,01 мл.
Реактивы
1. Стандартные растворы жирных кислот (муравьиной, уксусной, пропионовой, масляной, валерьяновой, капроновой, энантовой, каприловой, пеларгоновой) с содержанием 1 мг/мл метанола.
2. Силикагель марки СМ с зернами размером 0,5 — 2 мм.
Предварительно силикагель обрабатывают 50% (по объему) раствором соляной кислоты при 60 — 80 °C. Порции кислоты меняют до тех пор, пока она не будет бесцветной. Затем силикагель промывают водой и 2 — 3 раза — 50% (по объему) раствором азотной кислоты, отмывают горячей водой от ионов хлора и азотной кислоты, сушат при 105 °C и активируют в течение 30 мин. при 200 — 300 °C.
3. Метанол (метиловый спирт), обезвоженный металлическим магнием. Для этого в круглодонную колбу с обратным холодильником помещают 0,5 г йода, 5 г магния и 50 — 75 мл метанола. Смесь нагревают на водяной бане с помощью закрытой плитки в вытяжном шкафу. Горелки, расположенные поблизости, должны быть выключены, так как в результате реакции выделяется водород. (Если при этом не происходит бурного выделения водорода, добавляют еще 0,5 г йода.) Смесь нагревают до тех пор, пока весь магний не превратится в метилат магния. Затем прибавляют еще 900 мл метанола и смесь кипятят с обратным холодильником в течение 30 мин. Метанол отгоняют, предохраняя от влаги. Хранят в склянке с хорошо притертой пробкой.
4. Этанол 96°, обработанный едким кали; для этого к 100 мл спирта добавляют 20 г едкого кали, перемешивают и оставляют стоять 2 — 3 ч, после чего спирт отгоняют при 78°.
5. Серный эфир, для наркоза.
6. Едкий натр 0,05 н. водно-спиртовой (1:1) раствор.
7. Фенолфталеин 0,5% спиртовой раствор.
8. Едкий натр, 5% раствор в этиловом спирте.
9. Щелочной раствор гидроксиламина. К 10 мл 5% раствора гидроксиламина прибавляют 20 мл 5% раствора едкого натра и ставят на 5 — 10 мин. в сосуд со льдом. При этом выпадает обильный осадок хлористого натрия, который удаляют фильтрованием.
Данный раствор готовят в день употребления.
10. Соляная кислота, 2 н. раствор в этиловом спирте.
11. Хлорное железо, 0,5% раствор в этиловом спирте с добавлением 0,2 мл соляной кислоты (уд. вес 1,19).
12. Уксусная кислота, ледяная.
13. Муравьиная кислота.
14. Бутанол.
15. Бензол.
16. Щавелевая кислота, 1% раствор.
17. Серная кислота, уд. вес 1,84.
18. Натрий уксуснокислый, 0,5% раствор.
19. Йод, 0,1 М раствор в ледяной уксусной кислоте.
20. Сульфаниловая кислота, 0,5 г растворяют в 150 мл 30% уксусной кислоты.
21. ![]() -Нафтиламин, 0,1 г растворяют в 20 мл воды при нагревании до образования лиловых пятен на дне колбы. Раствор над осадком сливают в темную склянку и в нее вливают 110 мл 10% раствора уксусной кислоты.
-Нафтиламин, 0,1 г растворяют в 20 мл воды при нагревании до образования лиловых пятен на дне колбы. Раствор над осадком сливают в темную склянку и в нее вливают 110 мл 10% раствора уксусной кислоты.
22. Тиосульфат натрия, 0,1 н. раствор.
23. Хроматографическая бумага «Медленная», «Быстрая».
Отбор пробы
Для определения максимальной разовой концентрации воздух протягивают через два последовательно соединенных поглотительных прибора — Яворовской или НИИ гигиены имени Ф.Ф. Эрисмана, наполненные по 2 мл силикагеля. Протягивают 50 — 150 л исследуемого воздуха со скоростью 5 л/мин.
Ход анализа
1. Получение гидроксамовых кислот
а) Обработка проб.
После отбора пробы силикагель переносят в пробирку с притертой пробкой и заливают 5 мл этанола. Кислоты извлекают при комнатной температуре в течение 60 мин. при периодическом и интенсивном встряхивании.
Для анализа берут 3 мл прозрачного раствора, переносят в фарфоровый тигель и титруют 0,05 н. раствором едкого натра до слабо-розового окрашивания по фенолфталеину. Растворитель удаляют при 90 — 95 °C. Остаток натриевых солей жирных кислот подсушивают в эксикаторе или сушильном шкафу (80 °C). Сухой остаток солей смывают 1 мл метанола (дробными порциями), переносят в колориметрическую пробирку на 10 мл, прибавляют по 1 капле серной кислоты и оставляют на 30 мин. при комнатной температуре для образования эфиров. Через 30 мин. в каждую пробирку добавляют по 2 мл щелочного раствора гидроксиламина, заранее рассчитанное количество 5% раствора едкого натра для нейтрализации 1 капли серной кислоты и 5 мл эфира.
Смесь оставляют на 40 мин. при комнатной температуре для образования гидроксаматов. Через 40 мин. раствор нейтрализуют 2 н. раствором соляной кислоты по фенолфталеину до обесцвечивания малиновой окраски, содержимое взбалтывают и затем дают осесть осадку. Раствор над осадком сливают через складчатый фильтр, стараясь, чтобы на фильтр попало как можно меньше осадка. Затем снова к осадку в пробирке добавляют 1 — 1,5 мл эфира и повторяют извлечение гидроксамовых кислот. И так до 3 раз. Фильтрат переносят в выпарительную чашку и удаляют растворитель в вытяжном шкафу при комнатной температуре. Остаток смывают 0,3 мл метанола (дробными порциями) и переносят в круглодонную приборку с делением 0,3 мл, доводят этанолом до метки; 0,05 — 0,1 мл этого раствора используют для анализа кислот группы C1 — C4 и 0,05 — 0,1 мл для анализа кислот группы C5 — C9.
б) Приготовление «свидетелей».
Стандартные растворы кислот группы C1 — C4 смешивают по 0,2 мл каждой. Так же поступают со стандартными растворами кислот C5 — C9. Дальнейшая обработка стандартных растворов кислот проводится аналогично обработке проб воздуха.
После удаления растворителя полученные гидроксамовые кислоты растворяют в 1 мл метанола, что соответствует содержанию 200 мкг/мл каждой из кислот.
2. Разделение производных кислот
Для разделения производных кислот C1 — C4 используется хроматографическая бумага марок «Медленная» или «Быстрая» Ленинградской фабрики. На листе промытой бумаги проводят линию старта в 6,5 см от края, на которой намечают четыре точки с расстоянием 3,5 см друг от друга.
С помощью микропипетки в одну точку наносят 0,1 мл раствора смеси соответствующих «свидетелей» кислот группы C1 — C4, в три другие — пробы в количестве по 0,1 мл; при этом диаметр пятен на линии старта не должен превышать 5 — 6 мм.
С целью ускорения нанесения проб на бумагу следует пользоваться теплоэлектровентилятором.
Кислоты группы C5 — C9 делят на бумаге марки «Медленная».
После нанесения проб бумагу помещают в лодочку с подвижным растворителем, укрепленную в верхней части хроматографической камеры (предварительно насыщенную парами неподвижного и подвижного растворителей). Разделение идет нисходящим методом. Для разделения кислот C1 — C4 берут систему растворителей: бутанол, уксусную кислоту, воду в соотношении 4:1:5 помещают в делительную воронку, в которой при интенсивном встряхивании бутанол насыщается водой и уксусной кислотой. После разделения слоев нижний водный слой используют для насыщения камеры, для чего внутренние стенки ее покрывают фильтровальной бумагой, которая пропитывается растворителем. Концы бумаги не должны касаться друг друга, оставляя окно. Насыщать камеру следует не менее чем за 6 ч до начала анализа.
Верхний бутанольный слой из делительной воронки переносят в лодочку и используют в качестве подвижной фазы. Разделение кислот C1 — C4 идет в течение 18 — 20 ч. В качестве подвижной фазы при разделении кислот группы C5 — C9 используют бензол, насыщенный водой и муравьиной кислотой в соотношении 1:1:1. Разделение идет 5 — 6 ч. Разделение считается законченным, когда подвижный растворитель продвинется по бумаге на 25 — 30 см от линии старта. Далее хроматограмму извлекают из камеры, сушат в вытяжном шкафу при комнатной температуре в течение 30 — 60 мин. и проявляют.
3. Обнаружение производных кислот
Хроматограмму проявляют, опрыскивая из пульверизатора 0,5% раствором хлорного железа. Производные кислот проявляют в виде фиолетовых пятен на слабо-желтом фоне, которые расположены на хроматограмме последовательно в порядке увеличения числа углеродных атомов. Путем сравнения расположения на хроматограмме пятен проб с пятнами «свидетелей» делают выводы о наличии в пробах тех или иных кислот, так как производные одной и той же кислоты должны находиться на одинаковом расстоянии от линии старта (рис. 27).
Рис. 27. Хроматограмма карбоновых кислот гр. C1 — C4
(муравьиной, уксусной, пронионовой, масляной)
4. Количественное определение кислот группы
C1 — C9 методом визуальной оценки
Готовят растворы «свидетелей» с различным содержанием производных кислот. Для этого используют «свидетели», приготовленные согласно описанному выше способу. После получения гидроксамовых производных кислот растворитель испаряют, а сухие остатки производных растворяют в 1 мл этанола. Из полученных исходных растворов готовят серии разведений, как показано в табл. 87, 88.
Таблица 87
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ»
ДЛЯ ОПРЕДЕЛЕНИЯ КИСЛОТ C1 — C4
|
Исходный раствор гидроксамовых кислот, мл |
Этанол, мл |
Соответствующее количество каждой кислоты, мкг |
|
|
в 1 мл |
в 0,1 мл |
||
|
1,0 |
0 |
200,0 |
20,0 |
|
0,15 |
0,05 |
150,0 |
15,0 |
|
0,1 |
0,1 |
100,0 |
10,0 |
|
0,05 |
0,15 |
50,0 |
5,0 |
|
0,03 |
0,17 |
30,0 |
3,0 |
Таблица 88
ПРИГОТОВЛЕНИЕ СЕРИИ «СВИДЕТЕЛЕЙ» ДЛЯ ОПРЕДЕЛЕНИЯ
КИСЛОТ C5 — C9
|
Исходный раствор гидроксамовых кислот, мл |
Этанол, мл |
Соответствующее количество каждой кислоты, мкг |
|
|
в 1 мл |
в 0,1 мл |
||
|
0,5 |
0 |
400,0 |
40,0 |
|
0,1 |
0,1 |
200,0 |
20,0 |
|
0,1 |
0,3 |
100,0 |
10,0 |
|
0,05 |
0,35 |
50 |
5,0 |
Для определения кислот на лист хроматографической бумаги в разные точки наносят по 0,1 мл раствора «свидетелей». Затем «свидетели» подвергаются хроматографическому анализу в тех же условиях, что и пробы. На проявленных хроматограммах обнаруживаются пятна, различные по величине и интенсивности окраски и соответствующие 3 — 5 — 10 — 15 — 20 мкг каждой из кислот C1 — C4 и 5 — 10 — 20 — 40 мкг кислот C5 — C9.
Количественное определение кислот (после разделения их производных и проявления хроматограмм) методом визуальной оценки основано на том, что размер пятен и интенсивность окраски производных пропорциональны содержанию кислот. Сравнивая интенсивность окраски пятен проб с окраской «свидетелей», определяют содержание каждой кислоты в пробе.
5. Фотометрическое определение кислот C1 — C4
При фотометрическом определении содержания кислот в пробе предварительно готовят серию стандартов, как описано выше, т.е. подвергают хроматографическому разделению «свидетелей», соответствующих 3 — 5 — 10 — 15 — 20 мкг каждой из кислот C1 — C4. После проявления хроматограмм вырезают каждое пятно из бумаги, измельчают, помещают в пробирку и заливают 6 мл 0,5% раствора уксуснокислого натрия. Содержимое пробирки оставляют на 30 мин. при комнатной температуре, время от времени слегка встряхивают. Через 30 мин. 5 мл прозрачного раствора переносят в другую пробирку, добавляют 2 капли 0,1 М раствора йода и 0,3 мл раствора сульфаниловой кислоты.
Смесь помещают на 10 мин. в темное место. Через 10 мин. избыток йода связывают 0,1 н. раствором тиосульфата и добавляют 0,3 мл раствора ![]() -нафтиламина. Через 5 мин. величину оптической плотности раствора, окрашенного в малиновый цвет, измеряют при длине волны 520 нм.
-нафтиламина. Через 5 мин. величину оптической плотности раствора, окрашенного в малиновый цвет, измеряют при длине волны 520 нм.
Опыты повторяют 4 — 5 раз и по средним полученным данным строят градуировочный график зависимости оптической плотности элюатов от концентрации вещества.
Такие графики строят для каждой кислоты: муравьиной, масляной, уксусной, пропионовой.
Для определения содержания кислот в пробах после их хроматографического анализа последние обрабатывают так же, как и «свидетели». Величину оптической плотности окрашенного раствора проб измеряют на фотоколориметре и с помощью графиков находят количество кислоты в пробе.
Расчет см. на выше.
ЛИТЕРАТУРА
1. Абрамова Ю.В. Раздельное определение одноосновных карбоновых кислот в воздухе от C1 до C9 с помощью хроматографии на бумаге. Гиг. и сан., 1968, 6.
2. Абрамова Ю.В. К вопросу колориметрического определения жирных кислот группы C1 — C9 в воздухе. Гиг. и сан., 1969, 11.
3. Пименова З.М. Определение малеинового ангидрида. В кн.: М.С. Быховская, С.Л. Гинзбург и др. Методы определения вредных веществ в воздухе. «Медицина». М., 1966.
Глава XIII
АМИНОСОЕДИНЕНИЯ
ДИЭТИЛАМИН
Бесцветная жидкость с резким запахом, смешивается с водой, растворима в органических растворителях. Температура кипения 55,5 °C, плотность 0,710 (18 °C). Оказывает действие на центральную нервную систему, а также обладает раздражающими свойствами.
Предельно допустимые концентрации диэтиламина: максимальная разовая 0,05 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ ДИЭТИЛАМИНА С СЕРОУГЛЕРОДОМ [4]
Принцип определения
При взаимодействии диэтиламина с сероуглеродом и солями меди образуется диэтилдитиокарбамат меди, окрашенный в желто-зеленый цвет. Интенсивность окраски пропорциональна количеству диэтиламина.
Чувствительность определения — 0,5 мкг диэтиламина в анализируемом объеме раствора. Моноэтиламин до 50 мкг, а также триэтиламины и аммиак определению не мешают.
Реактивы
1. Поглотительный раствор: этиловый спирт, 96% ректификат.
2. Стандартный раствор диэтиламина. Исходный стандартный раствор готовят следующим образом. В мерную колбу емкостью 25 — 50 мл вливают 10 — 15 мл этилового спирта, взвешивают, вносят несколько капель диэтиламина и снова взвешивают. Объем жидкости в колбе доводят до метки этиловым спиртом и хорошо перемешивают. По разности в весе определяют количество диэтиламина и вычисляют его содержание в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл диэтиламина готовят соответствующим разведением исходного раствора спиртом.
3. Сероуглерод, 10% спиртовой раствор (по объему), 1 мл сероуглерода вносят в небольшое количество спирта в пробирку с притертой пробкой и доводят объем до 10 мл спиртом.
4. Ацетат меди, спиртовой раствор, 10 мг соли растворяют в 0,5 мл воды и добавляют 9,5 мл этилового спирта.
5. Серная кислота, уд. вес 1,84.
6. Аммиак, спиртовой раствор, 1 мл 25% раствора аммиака разбавляют 9 мл этилового спирта. К 4 мл воды в конической колбе добавляют 1 мл спиртового раствора аммиака и оттитровывают 0,1 н. раствором соляной кислоты в присутствии индикатора метилового красного. Контрольный опыт обязателен. 1 мл 0,1 н. раствора соляной кислоты соответствует 1,7 мг аммиака. Готовят раствор, содержащий 7 — 10 мг/мл.
Растворы сероуглерода, ацетата меди и аммиака устойчивы в течение 2 — 3 дней.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 2 мл 96% этилового спирта. Продолжительность отбора 20 — 30 мин. Поглотительные приборы следует охлаждать, при испарении доводить до первоначального объема.
Ход анализа
1 мл пробы вносят в колориметрическую пробирку. Одновременно готовят шкалу стандартов (табл. 89).
Таблица 89
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
|
Этиловый спирт, мл |
1 |
0,95 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
|
Содержание диэтиламина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
В каждую пробирку шкалы и пробы прибавляют 0,1 мл спиртового раствора аммиака, 0,1 мл 10% спиртового раствора сероуглерода и 0,1 мл спиртового раствора ацетата меди и оставляют на 6 — 7 мин. По истечении этого времени добавляют по 0,25 мл серной кислоты уд. веса 1,84. После добавления каждого реактива раствор перемешивают. Интенсивность окраски пробы сравнивают со шкалой стандартов.
Расчет см. выше.
ТРИЭТИЛАМИН
Бесцветная жидкость с аммиачным запахом. Плотность 0,728 (20°), температура кипения 89,5°.
Триэтиламин очень хорошо растворим в органических растворителях.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,14 мг/м3.
ОПРЕДЕЛЕНИЕ ТРИЭТИЛАМИНА С БРОМФЕНОЛОВЫМ СИНИМ [4]
Принцип определения
При взаимодействии триэтиламина с бромфеноловым синим образуется соединение, окрашенное в желтый цвет.
Чувствительность определения — 1 мкг триэтиламина в анализируемом объеме раствора. Моноэтиламин, диэтиламин, аммиак определению не мешают. Другие третичные амины мешают определению.
Реактивы
1. Поглотительный раствор: 0,01 н. раствор соляной кислоты.
2. Стандартный раствор триэтиламина. Исходный раствор готовят в мерной колбе емкостью 25 мл, в которую вносят 2 — 3 мл 0,01 н. раствора соляной кислоты, взвешивают, вносят 2 — 3 капли триэтиламина и снова взвешивают. Объем жидкости доводят до метки 0,01 н. раствором соляной кислоты и рассчитывают содержание триэтиламина в 1 мл. Рабочий стандартный раствор с содержанием 10 мкг/мл готовят соответствующим разбавлением исходного раствора 0,01 н. раствором соляной кислоты.
3. Раствор бромфенолового синего. 0,1 г реактива растворяют в 10 мл этилового спирта и разбавляют водой до объема 100 мл.
4. Ацетатный буферный раствор с pH 3,5 — 4,5. 50 мл 1 н. раствора ацетата натрия смешивают с 40 мл 1 н. раствора соляной кислоты и доводят водой до объема 250 мл.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор с пористой пластинкой N 1, содержащий 6 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,3 — 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром, содержащий 6 мл поглотительного раствора непрерывно в течение суток. Перемешивают, охлаждают в воде со льдом. Добавляют 4 мл хлороформа, вновь охлаждают.
Ход анализа
К 5 мл пробы добавляют 1 мл ацетатного буферного раствора и 5 мл бромфенолового синего. Перемешивают и охлаждают в течение 5 мин. в воде со льдом. Добавляют 4 мл хлороформа, вновь охлаждают 5 мин. Смесь помещают в делительную воронку, перемешивают в течение 15 мин. Слой хлороформа переносят в кювету с толщиной слоя 10 мм, фотометрируют при 420 нм по сравнению с контролем, который готовят аналогично пробе.
Содержание триэтиламина определяют по калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 90).
Таблица 90
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,0 |
|
Содержание триэтиламина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой можно пользоваться для визуального определения, ее готовят одновременно с пробами.
Расчет см. выше.
ГЕКСАМЕТИЛЕНДИАМИН (ГМД)
|
NH2(CH2)6NH2 |
Мол. вес 116,2 |
Твердое вещество, обладающее резким неприятным запахом, легко растворимое в воде и органических растворителях. Температура плавления 39 °C, температура кипения 196 °C. ГМД легко возгоняется, является сильным основанием. Оказывает действие на нервную систему и вызывает изменения в крови.
Предельно допустимые концентрации гексаметилендиамина: максимальная разовая 0,001 мг/м3, среднесуточная 0,001 мг/м3.
ОПРЕДЕЛЕНИЕ ГЕКСАМЕТИЛЕНДИАМИНА
С 2,4-ДИНИТРОХЛОРБЕНЗОЛОМ [7]
Принцип определения
При взаимодействии гексаметилендиамина с 2,4-динитрохлорбензолом образуется соединение, окрашенное в желтый цвет. Интенсивность окраски пропорциональна количеству гексаметилендиамина.
Чувствительность определения — 0,3 мкг в анализируемом объеме раствора.
Определению мешает аммиак с 5 мг в анализируемом объеме, а также первичные, вторичные и некоторые третичные амины.
Реактивы
1. Поглотительный раствор: серная кислота, 0,1 н. раствор.
2. Стандартные растворы ГМД.
Исходный раствор с содержанием 100 мкг/мл готовят растворением 0,05 г ГМД (бесцветного) в 500 мл 0,1 н. раствора серной кислоты. Рабочий стандартный раствор, содержащий 1 мкг/мл ГМД, готовят разведением исходного раствора в 100 раз 0,1 н. раствором серной кислоты.
3. Карбонат натрия (Na2CO3·10H2O), 36% раствор. Готовят при нагревании на водяной бане.
4. 2,4-Динитрохлорбензол, 5% спиртовой раствор. Готовят при нагревании на водяной бане.
5. Соляная кислота, уд. вес 1,19.
6. Хлороформ.
Отбор пробы
Воздух со скоростью 2 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1 (диаметр фильтра 20 — 25 мм), содержащих по 10 мл 0,1 н. раствора серной кислоты.
Ход анализа
Для анализа из каждого поглотительного прибора берут 9 мл пробы и вносят в конические колбы с притертыми пробками емкостью 25 — 30 мл; в таких же колбах готовят шкалу стандартов (табл. 91).
Таблица 91
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Рабочий стандартный раствор, мл |
0 |
0,3 |
0,6 |
1,2 |
2,4 |
3,6 |
|
Поглотительный раствор, мл |
9 |
8,7 |
8,4 |
7,8 |
6,6 |
5,4 |
|
Содержание ГМД, мкг |
0 |
0,3 |
0,6 |
1,2 |
2,4 |
3,6 |
Во все колбы шкалы и проб вливают по 0,6 мл 36% раствора карбоната натрия и по 1,2 мл 5% раствора динитрохлорбензола. Колбы без пробок помещают в кипящую водяную баню на 30 мин., закрепив их в штативе. Колбы охлаждают и вливают по 0,3 мл соляной кислоты уд. веса 1,19 и по 0,5 мл (точно) хлороформа. Колбы закрывают пробками и ставят на 20 мин. в аппарат для встряхивания и полного экстрагирования. После экстрагирования растворы переливают в конические центрифужные пробирки и колориметрируют. Шкала в закрытых пробирках устройства в течение 2 нед.
Расчет см. выше.
АРОМАТИЧЕСКИЕ АМИНЫ
АНИЛИН
Бесцветная маслянистая жидкость с характерным запахом, быстро темнеющая на свету, плотность при 20 °C 1,022, температура кипения 184,4 °C. Анилин хорошо растворим в эфире, спирте, ацетоне, бензоле, хлороформе и других органических растворителях. При 20 °C 3,4 г анилина растворяются в 100 мл воды. Анилин обладает основными свойствами, с кислотами образует соли.
Анилин действует на центральную нервную систему и является ядом крови.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3, среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ АНИЛИНА ПО ИНДОФЕНОЛУ [1]
Принцип определения
При взаимодействии анилина и фенола с хлорамином в щелочной среде образуется индофенол, который окрашивает раствор в синий цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Ароматические амины и аммиак мешают определению.
Реактивы
1. Поглотительный раствор: 0,01 н. раствор серной кислоты готовят разбавлением 0,1 н. серной кислоты в 10 раз.
2. Силикагель АСМ, КСМ, КСК с зернами величиной 0,25 — 2 мм, промытый водой при кипячении, высушенный при 105° и активированный при 200° в течение 30 мин., проверенный на отсутствие анилина.
3. Стандартный раствор анилина. Исходный стандартный раствор готовят растворением точной навески анилина в 0,01 н. растворе серной кислоты. Рассчитывают содержание анилина в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл готовят из исходного соответствующим разбавлением его 0,01 н. раствором серной кислоты.
4. Хлорамин, 4% раствор, растворяют в воде, нагретой до 40 — 50° и фильтруют.
5. Фенол, 3% раствор. Готовят из бесцветного свежеперегнанного фенола.
6. Натр едкий, 2% и 0,1 н. растворы.
7. Серная кислота, 0,1 н. раствор.
Отбор пробы
а) Для определения разовой концентрации протягивают воздух со скоростью 1 л/мин. через поглотительный прибор с пористым фильтром N 1, содержащий 4 мл поглотительного раствора. Отбор проб можно проводить в «кипящий» слой силикагеля. Для этого 3 мл силикагеля помещают в два последовательно присоединенных поглотительных прибора — Яворовской и Института имени Ф.Ф. Эрисмана, соединенных с поглотителем для улавливания переброшенных частиц. Отбирают пробу со скоростью до 10 л/мин. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,3 л/мин. протягивают через поглотительный прибор, содержащий 4 мл поглотительного раствора. При наполнении прибора отмечают уровень жидкости, который периодически добавляют водой до первоначального. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Для отбора пробы также используют и силикагель, скорость отбора до 5 л/мин.
Ход анализа
Силикагель переносят отдельно из каждого поглотительного прибора в пробирки, переброшенные зерна присоединяют ко второму прибору.
Добавляют по 3 мл этилового спирта, нагревают до 50° на водяной бане в течение 20 мин., вносят по 3 мл дистиллированной воды, перемешивают. По охлаждении к пробе прибавляют по 0,5 мл 4% раствора хлорамина, 0,5 мл 3% раствора фенола и по 0,5 мл 2% раствора едкого натра. Содержимое пробирок встряхивают и интенсивность окраски пробы сравнивают со шкалой стандартов (табл. 92).
Таблица 92
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
3 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
|
Содержание анилина, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе.
Расчет см. выше.
При отборе проб в жидкую среду 3 мл пробы переносят в пробирку, вносят 0,3 мл 0,1 н. раствора щелочи для нейтрализации, 0,5 мл 4% раствора хлорамина, раствор встряхивают, добавляют 0,5 мл 3% раствора фенола и 0,2 мл 2% раствора щелочи. Содержимое пробирки встряхивают и через 10 — 20 мин. измеряют оптическую плотность при длине волны 660 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание вещества в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов, но без добавления силикагеля. Все пробирки шкалы обрабатывают аналогично пробе и строят график.
Расчет см. выше.
МОНОМЕТИЛАНИЛИН
Прозрачная бесцветная жидкость, температура кипения 197,7 °C, плотность при 20 °C 0,986. Метиланилин в воде не растворим, растворим в органических растворителях.
На организм действует так же, как анилин.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,04 мг/м3.
ОПРЕДЕЛЕНИЕ МОНОМЕТИЛАНИЛИНА С ХЛОРАМИНОМ [3]
Принцип определения
При взаимодействии метиланилина с хлорамином и фенолом в щелочной среде образуется соединение, которое окрашивает раствор в синий цвет.
Чувствительность определения — 1 мкг в анализируемом объеме раствора. Другие первичные и вторичные ароматические амины и аммиак мешают определению.
Реактивы
1. Поглотительный раствор, 2% раствор этанола.
2. Стандартный раствор метиланилина: готовят исходный стандартный раствор в мерной колбе емкостью 50 мл, в которую наливают 20 мл 40% раствора этанола, колбу взвешивают, в нее вносят 2 — 3 капли метиланилина и снова взвешивают. Раствор в колбе взбалтывают и объем его доводят до метки 40% раствором этанола. По разности двух взвешиваний устанавливают количество метиланилина в колбе и вычисляют содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в которую вносят такое количество исходного раствора, которое содержит 500 мкг метиланилина. Раствор доводят до метки 2% раствором этанола. Применяют свежеприготовленный раствор.
3. Этанол 96°, 2 и 40% растворы.
4. Хлорамин Б, 6% раствор. После растворения и отстаивания раствор фильтруют.
5. Фенол, 6% раствор.
6. Тиосульфат натрия, 0,1 н. раствор.
7. Едкий натр, 6% раствор.
Отбор пробы
Воздух протягивают со скоростью 0,5 л/мин. через два поглотительных прибора с пористым фильтром N 1, наполненных по 5 мл поглотительного раствора.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. По 3 мл пробы из каждого поглотительного прибора переносят в пробирки, вливают по 0,5 мл хлорамина, раствор встряхивают и через 5 мин. добавляют 0,1 мл раствора тиосульфата, 0,1 мл щелочи и 0,4 мл раствора фенола. После каждого добавления растворы встряхивают. Через 20 мин. измеряют оптическую плотность раствора при длине волны 620 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание метиланилина в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения графика готовят шкалу стандартов (табл. 93).
Таблица 93
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
3 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
1,0 |
|
Содержание метиланилина, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
20 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ДИМЕТИЛАНИЛИН
|
C6H5N(CH3)2 |
Мол. вес 121,19 |
Бесцветная жидкость с неприятным запахом, температура кипения 193 °C, плотность при 20 °C 0,956, мало растворим в воде, но хорошо растворим в этаноле, эфире и кислотах.
Диметиланилин действует на организм подобно анилину.
Предельно допустимые концентрации: максимальная разовая, среднесуточная 0,0055 мг/м3.
ОПРЕДЕЛЕНИЕ ДИМЕТИЛАНИЛИНА С СУЛЬФАНИЛОВОЙ КИСЛОТОЙ [6]
Принцип определения
При взаимодействии диметиланилина с диазотированной сульфаниловой кислотой образуется соединение, окрашивающее раствор в розовый цвет, по интенсивности которого определяют содержание диметиланилина.
Чувствительность определения — 0,4 мкг в анализируемом объеме.
Первичные амины мешают определению.
Реактивы
1. Поглотительный раствор, 0,01 н. раствор серной кислоты.
2. Стандартный раствор диметиланилина. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 5 — 10 мл поглотительного раствора, колбу взвешивают, вносят в нее 2 — 3 капли диметиланилина и вновь взвешивают, объем жидкости в колбе доводят до метки поглотительным раствором. По разности двух взвешиваний устанавливают количество диметиланилина в колбе и рассчитывают содержание его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 1 мкг/мл получают в мерной колбе емкостью 100 мл, в нее вносят такое количество исходного раствора, которое содержит 100 мкг диметиланилина. Объем жидкости в колбе доводят до метки поглотительным раствором.
3. Нитрит натрия, 0,2% раствор.
4. Едкий натр, 0,1 н. раствор.
5. Соляная кислота, 1 н. раствор.
6. Составной реактив: 0,2 г сульфаниловой кислоты растворяют в 20 мл 0,1 н. раствора едкого натра и добавляют 20 мл 0,2% раствора нитрита натрия. Объем раствора доводят до 100 мл водой. Раствор готовят перед применением.
7. Сульфаниловая кислота.
Отбор пробы
а) Для определения разовой концентрации пропускают воздух со скоростью 1 л/мин. через поглотительный прибор с пористым фильтром N 1, наполненный 3 мл поглотительного раствора. Продолжительность отбора более часа.
б) Для определения среднесуточной концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, наполненный 3 мл поглотительного раствора. Отбор проводят в течение суток. Допустимо проводить отбор 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор. При наполнении прибора отмечают уровень жидкости, который периодически доводят до первоначального.
Допустимо отбор проводить 6 — 12 раз с перерывами 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В колориметрическую пробирку помещают 2 мл пробы. В пробирку прибавляют 2 мл составного реактива и 4 капли 1 н. раствора соляной кислоты. Раствор встряхивают и через 15 мин. интенсивность окраски пробы сравнивают со шкалой стандартов (табл. 94).
Таблица 94
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
|
Поглотительный раствор, мл |
2 |
1,6 |
1,4 |
1,2 |
1,0 |
0,5 |
0 |
|
Содержание диметиланилина, мкг |
0 |
0,4 |
0,6 |
0,8 |
1,0 |
1,5 |
2,0 |
Все пробирки шкалы обрабатывают аналогично пробе и одновременно с ней.
Расчет см. выше.
МЕЗИДИН
|
(CH3)3C6H2NH |
Мол. вес 135,21 |
Маслообразная прозрачная жидкость темно-коричневого цвета с резким запахом, температура кипения 233 °C, плотность при 20 °C 0,963. Не растворим в воде, растворим в уксусной кислоте, этаноле, эфире и других органических растворителях.
Токсическое действие на организм человека сходно с действием анилина.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,003 мг/м3.
ОПРЕДЕЛЕНИЕ МЕЗИДИНА ПО ОБРАЗОВАНИЮ ДИАЗОСОЕДИНЕНИЯ [8]
Принцип определения
При взаимодействии мезидина и нитрита натрия в кислой среде образуется диазосоединение, при сочетании последнего с ![]() -нафтолом в карбонатной среде раствор окрашивается в розово-красный цвет, по интенсивности которого определяют содержание мезидина.
-нафтолом в карбонатной среде раствор окрашивается в розово-красный цвет, по интенсивности которого определяют содержание мезидина.
Чувствительность определения — 0,1 мкг в анализируемом объеме.
Анилин мешает определению.
Реактивы
1. Поглотительный раствор 0,01 н. раствор серной кислоты.
2. Стандартный раствор мезидина: готовят исходный раствор в мерной колбе емкостью 50 мл, в которую вливают 10 — 20 мл 0,1 н. раствора серной кислоты. Колбу взвешивают, в нее вносят 2 — 3 капли мезидина и вновь взвешивают. Объем жидкости в колбе доводят до метки 0,1 н. раствором серной кислоты. По разности двух взвешиваний устанавливают содержание мезидина в колбе и вычисляют количество его в 1 мл раствора.
Рабочий стандартный раствор с содержанием 1 мкг/мл готовят соответствующим разведением исходного раствора.
3. Карбонат натрия (безводный) 5 н. раствор, свежеприготовленный.
4. Нитрит натрия, 2% раствор.
5. Серная кислота, 0,1 н. и 0,01 н. растворы.
6. ![]() -Нафтол, 5% спиртовой раствор.
-Нафтол, 5% спиртовой раствор.
Отбор пробы
а) Для определения максимально разовой концентрации воздух протягивают со скоростью 1 л/мин. через два соединенных поглотительных прибора с пористым фильтром N 1, наполненных 3 мл 0,01 н. раствором серной кислоты каждый. Продолжительность отбора 30 мин.
б) Для определения среднесуточной концентрации непрерывно в течение суток воздух со скоростью 0,5 л/мин. протягивают через указанные выше приборы. При наполнении приборов раствором отмечают уровень жидкости, который периодически добавляют водой до первоначального.
Допустимо проводить отбор с перерывами на 2 — 4 ч в те же приборы.
Ход анализа
Каждый поглотительный прибор анализируют отдельно. В колориметрическую пробирку переносят 2 мл пробы и прибавляют 0,2 мл нитрита натрия, 0,3 мл карбоната натрия и 0,2 мл раствора ![]() -нафтола. Пробирку встряхивают после каждого добавления раствора.
-нафтола. Пробирку встряхивают после каждого добавления раствора.
Через 5 мин. окрашенный раствор пробы сравнивают со шкалой стандартов, которую готовят одновременно и аналогично с пробой (табл. 95).
Расчет см. выше.
Таблица 95
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
Номер пробирки |
|||
|
1 |
2 |
3 |
4 |
||
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
|
Поглотительный раствор, мл |
2,0 |
1,9 |
1,7 |
1,5 |
1,0 |
|
Содержание мезидина, мкг |
0 |
0,1 |
0,3 |
0,5 |
1,0 |
ПИРИДИН
Жидкость без цвета, с резким запахом. Растворим в воде, спирте, эфире, бензоле.
Плотность 0,9878 (20 °C), плотность паров по отношению к воздуху 2,75. Температура кипения 115,58 °C.
Продукты замещения атомов водорода в ядре пиридина носят название метилпроизводных; однозамещенных — пиколинов, дву- и тризамещенных — лутидинов, коллидинов. Все эти соединения относятся к пиридиновым основаниям. Пиридин и пиридиновые основания токсичны. Поражают нервную систему, вызывают раздражение слизистых оболочек дыхательных путей, глаз. Поражают печень, почки, действуют на желудочно-кишечный тракт.
Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,08 мг/м3.
ОПРЕДЕЛЕНИЕ ПИРИДИНА С ХЛОРАМИНОМ И РОДАНИДОМ КАЛИЯ [5]
Принцип определения
При взаимодействии пиридина с хлорамином, роданидом калия или аммония и барбитуровой кислотой при pH 4,5 — 6,5 образуется соединение, окрашенное в розовый цвет.
Чувствительность — 0,25 мкг в определяемом объеме раствора.
Определению мешают пиридиновые основания.
Реактивы
1. Поглотительный раствор. Серная кислота 0,10 н. раствор.
2. Стандартный раствор пиридина. Чистый пиридин высушивают над прокаленной окисью кальция. Для этого в сухую склянку с притертой пробкой насыпают окись кальция и наливают пиридин. Окись кальция расплывается. Пиридин сливают в другую склянку, в которую снова вносят свежепрокаленную окись кальция. Повторяют операции до тех пор, пока окись кальция перестанет расплываться. Пиридин сливают и перегоняют. Собирают фракцию с температурой кипения 115,6 °C.
В мерную колбу емкостью 100 мл вносят около 5 мл 0,1 н. раствора серной кислоты, взвешивают. Вносят 0,1 мл перегнанного пиридина и снова взвешивают, доводят объем до метки. Получают исходный раствор с содержанием около 1 мг/мл пиридина.
Рабочий стандартный раствор с содержанием 5 мкг/мл готовят разбавлением исходного 0,1 н. раствором серной кислоты.
3. Натр едкий, 0,1 н. раствор.
4. Роданид аммония или калия, 1% раствор.
5. Хлорамин «В», 8% раствор.
6. Барбитуровая кислота, 1% раствор.
Навеску барбитуровой кислоты растворяют в воде, нагретой до 50 — 60 °C.
7. Фосфат калия 0,1 М раствор.
Навеску однозамещенного фосфата калия 13,613 г растворяют в небольшом объеме воды в мерной колбе емкостью 1 л. После растворения объем доводят до метки.
8. Фосфатнощелочной буфер, pH 6,0 — 6,2 К 50 мл 0,1 М раствора фосфата калия добавляют 5,7 мл 0,1 н. раствора едкого натра и объем доводят до 100 мл.
9. Универсальный индикатор, раствор.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,2 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористой пластинкой N 1 (малая модель), содержащих по 2 мл 0,1 н. раствора серной кислоты.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,1 л/мин. протягивают через два последовательно соединенных поглотительных прибора с пористой пластинкой N 1 (малая модель), содержащих по 2 мл 0,1 н. раствора серной кислоты, отмечая уровень жидкости.
Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня.
Допустимо проводить отбор проб с перерывами в 2 — 4 ч в одну и ту же систему поглотителей.
Ход анализа
По 1 мл пробы переносят в пробирки.
Предварительно устанавливают количество щелочи, которое необходимо добавить в пробу для создания соответствующего pH. Для этого к 1 мл 0,1 н. раствора серной кислоты добавляют 1 мл 1% раствора роданида калия или аммония и 1 мл 8% раствора хлорамина. Через 10 мин. вносят 3 мл 1% раствора барбитуровой кислоты и 4 — 5 капель универсального индикатора и титруют 0,1 н. раствором щелочи до желтой окраски (pH 4,5 — 6,5), получают значение (а).
К пробе добавляют по 1 мл 1% раствора роданида калия или аммония, 1 мл 8% раствора хлорамина, тщательно перемешивают и через 10 мин. вносят по 3 мл 1% раствора барбитуровой кислоты и (а) мл 0,1 н. раствора щелочи, количество которой было установлено. Объем пробы доводят до объема 7 мл путем добавления фосфатнощелочного буфера.
Пробирки помещают на водяную баню и выдерживают в течение 30 — 35 мин. при 35 — 45 °C.
После охлаждения раствор фотометрируют в кюветах с толщиной слоя 1 см при длине волны 584 нм по сравнению с контролем. Содержание пиридина в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 96).
Таблица 96
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
1 |
0,95 |
0,9 |
0,8 |
0,6 |
0,4 |
0,2 |
0 |
|
Содержание пиридина, мкг |
0 |
0,25 |
0,5 |
1,0 |
2,0 |
3,0 |
4,0 |
5,0 |
Все пробирки шкалы обрабатывают аналогично пробе.
Шкалой стандартов можно пользоваться для визуального определения, ее готовят одновременно с пробами.
Расчет см. выше.
ДИМЕТИЛФОРМАМИД
Бесцветная жидкость со специфическим запахом, на свету не разлагается. Температура кипения 153 °C, плотность 0,96 (25°), упругость пара 3,7 мм рт. ст. Смешивается в любых отношениях с водой, спиртом, простыми и сложными эфирами. Оказывает раздражающее действие на верхние дыхательные пути, глаза и кожу. Предельно допустимые концентрации: максимальная разовая и среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ДИМЕТИЛФОРМАМИДА С ДИНИТРОХЛОРБЕНЗОЛОМ [2]
Принцип определения
Метод основан на гидролизе диметилформамида щелочью и взаимодействием продукта гидролиза — диметиламина с 2,4-динитрохлорбензолом. Образуется соединение, окрашенное в желтый цвет. Интенсивность окраски пропорциональна количеству диметилформамида.
Чувствительность определения — 0,5 мкг в анализируемом объеме.
Определению мешают амины.
Реактивы
1. Поглотительный раствор — соляная кислота, 0,05 н. раствор.
2. Стандартные растворы диметилформамида. Исходный стандартный раствор готовят следующим образом. В мерную колбу емкостью 25 — 50 мл вливают 10 — 15 мл 0,05 н. раствора соляной кислоты, взвешивают, вносят 2 — 3 капли диметилформамида и снова взвешивают. Объем жидкости в колбе доводят до метки 0,05 н. раствором соляной кислоты и хорошо перемешивают. По разности в весе определяют количество диметилформамида и вычисляют его содержание в 1 мл раствора. Рабочий стандартный раствор с содержанием 10 мкг/мл диметилформамида готовят соответствующим разведением исходного раствора 0,05 н. раствором соляной кислоты.
3. Соляная кислота, 0,01 н., 0,05 н. и 25% растворы.
4. 20% раствор едкого натра или едкого кали.
5. Карбонат натрия кристаллический, 8% раствор.
6. 2,4-Динитрохлорбензол, 1% спиртовой раствор. Готовят при нагревании на водяной бане.
7. Хлороформ.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через поглотительный прибор Зайцева, содержащий 5 мл 0,05 н. раствора соляной кислоты. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,25 л/мин. протягивают через поглотительный прибор Зайцева, содержащий 5 мл 0,05 н. раствора соляной кислоты. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо отбор проводить 6 — 12 раз с перерывами в 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В поглотительный прибор с пробой для гидролиза диметилформамида вливают 1 мл 20% раствора едкого натра. Этот поглотительный прибор соединяют с другим поглотительным прибором Зайцева, наполненным 4 мл 0,01 н. раствора соляной кислоты. Поглотительный прибор с пробой помещают в кипящую водяную баню точно на 5 мин., баня должна быть закрытой, и в это время через поглотительные приборы пропускают воздух со скоростью 0,1 л/мин. Образовавшийся диметиламин поглощается 0,01 н. раствором соляной кислоты. Для исключения возможности переброса жидкости поглотительный прибор с продуктом гидролиза быстро отъединяют от всей системы при включенном аспираторе. Для анализа берут в колориметрическую пробирку 2 мл раствора. Одновременно готовят шкалу стандартов, для этого в поглотительные приборы Зайцева через длинную трубку вносят рабочий стандартный раствор в количестве 0,1; 0,2; 0,8; 1,2; 1,6; 2 мл и доводят до 4 мл 0,05 н. раствором соляной кислоты. Кислоту вливают также через длинную трубку поглотителя, чтобы смыть с ее стенок диметилформамид. Во все поглотительные приборы вливают по 1 мл 20% раствора щелочи и обрабатывают так же, как и пробу. Для составления шкалы стандартов после гидролиза берут в колориметрические пробирки половину, т.е. 2 мл раствора, что соответствует следующим количествам диметилформамида (табл. 97).
Таблица 97
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Содержание диметилформамида, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Контрольный опыт ставят так же, как и пробу. Во все пробирки шкалы и проб прибавляют по 0,2 мл 8% раствора карбоната натрия, растворы встряхивают и к ним добавляют по 0,2 мл 1% раствора динитрохлорбензола. Все пробирки помещают на 5 мин. в кипящую водяную баню. После охлаждения в пробирки прибавляют по 0,2 мл 25% раствора соляной кислоты и после взбалтывания растворов добавляют по 0,5 мл хлороформа. Растворы снова встряхивают и через 15 — 20 мин. сравнивают желтую окраску проб со шкалой стандартов.
Расчет см. выше.
ЛИТЕРАТУРА
1. Алексеева М.В. Определение анилина в воздухе. В сб.: Предельно допустимые концентрации атмосферных загрязнений. М., «Медгиз», 1963, в. 7.
2. Алексеева М.В. Определение диметилформамида. В кн.: Определение атмосферных загрязнений. М., «Медицина», 1963.
3. Беляков А.А., Горбылева Н.В. Определение микрограммовых количеств диметиланилина, анилина и метиланилина в смеси. Аналитическая химия, 1957, т. 12, 4.
4. Виноградова В.А., Помазова Е.Н. Определение низших алифатических аминов в воздухе. Всесоюзная конференция по новым методам определения микроконцентраций токсических веществ в воздухе и других средах. Харьков, 1972.
5. Горская Р.В., Ярым-Агаева Н.Т. Фотометрическое определение малых количеств пиридина. Аналитическая химия, 1965, 6.
6. Зильберг Л.А. К вопросу об определении диметиланилина в воздухе производственных помещений. Гиг. и сан., 1957, 6.
7. Кузнецов В.И., Пименова З.М. и др. Определение гексаметилендиамина. В кн.: Определение вредных веществ в воздухе. М., Медгиз, 1957.
8. Полежаев Н.Г., Кузнецова Л.В. Новый колориметрический метод определения мезидина в воздухе. Гиг. и сан., 1967, 4.
Глава XIV
РАЗНЫЕ ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
КАПРОЛАКТАМ
Бесцветное кристаллическое вещество, растворимое в воде и органических растворителях, температура плавления 68 — 70 °C, температура кипения 139 — 140 °C (12 мм рт. ст.).
Капролактам вызывает головные боли, раздражительность, нарушение сна, при действии на кожу может вызывать дерматиты.
Предельно допустимые концентрации капролактама (пары, аэрозоль): максимальная разовая и среднесуточная 0,06 мг/м3.
ОПРЕДЕЛЕНИЕ КАПРОЛАКТАМА С ГИДРОКСИЛАМИНОМ [7]
Принцип определения
При воздействии капролактама с гидроксиламином в щелочной среде образуется гидроксамовая кислота, которую определяют колориметрически по реакции с хлорным железом.
Чувствительность определения — 50 мкг капролактама в анализируемом объеме раствора.
Определению мешают сложные эфиры карбоновых кислот и другие вещества, реагирующие с гидроксиламином с образованием гидроксамовой кислоты.
Определению не мешают циклогексанон, циклогексанол, циклогексаноноксим, нитроциклогексан, бензол, аммиак, сернистый газ и серный ангидрид.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор капролактама. Исходный стандартный раствор, содержащий 1000 мкг/мл капролактама, готовят путем растворения 0,1 г капролактама в 100 мл воды.
3. Гидроксиламин солянокислый, 3 М раствор.
В 100 мл воды растворяют 21 г солянокислого гидроксиламина. Раствор должен быть свежеприготовленный.
4. Хлорид железа, 10% раствор. Растворяют 10 г хлорида окисного железа в 80 мл воды и добавляют 15 мл 2 н. раствора соляной кислоты. Раствор годен к употреблению в течение недели.
5. Натр едкий, 1 н. раствор.
6. Соляная кислота, 2 н. раствор.
7. Контрольный раствор. К 10 мл воды добавляют 4 мл раствора гидроксиламина и 1 мл раствора едкого натра. Контрольный раствор готовят перед употреблением.
Отбор пробы
а) Для определения максимальной разовой концентрации исследуемый воздух протягивают со скоростью 10 л/мин. через фильтр АФА-ХС, укрепленный в патроне, и через поглотительный прибор Гернет, содержащий 8 мл дистиллированной воды, соединенный последовательно с патроном.
б) Для определения среднесуточной концентрации воздух со скоростью 2 — 5 л/мин. непрерывно в течение суток протягивают через фильтр АФА-ХС, укрепленный в патроне, и поглотительный прибор Гернет, содержащий 8 мл дистиллированной воды, отмечая уровень жидкости и периодически добавляя воду до первоначального объема. Допустимо отбор проводить на один и тот же фильтр и поглотитель с перерывами в 2 или 4 ч.
Ход анализа
Фильтр переносят в стакан, промывают 5 мл воды, отжимая стеклянной палочкой. Раствор сливают в круглодонную колбу емкостью 75 — 100 мл, добавляют 2 мл раствора солянокислого гидроксиламина и 0,5 мл 1 н. раствора едкого натра. Затем присоединяют обратный холодильник, раствор нагревают и кипятят точно 30 мин. от начала закипания. Раствор охлаждают до комнатной температуры, сливают в пробирку, колбу ополаскивают 0,5 мл контрольного раствора, который сливают в ту же пробирку.
Для анализа берут в колориметрическую пробирку 3 мл пробы. Из поглотительного прибора Гернет берут 5 мл пробы и обрабатывают, как описано выше.
Для составления шкалы стандартов проводят гидролиз капролактама для перевода его в гидроксамовую кислоту. В день анализа в колбу с обратным холодильником вносят 10 мл рабочего стандартного раствора, добавляют 4 мл гидроксиламина, 1 мл раствора едкого натра, кипятят точно 30 мин. от начала закипания, охлаждают. Доводят объем до 15 мл и получают рабочий стандартный раствор с содержанием 667 мкг/мл капролактама. Рабочий стандартный раствор наносят на фильтры, применяемые при отборе, и готовят шкалу стандартов (табл. 98).
Таблица 98
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Раствор гидроксамовой кислоты, полученной после гидролиза капролактама, мл |
0 |
0,075 |
0,12 |
0,15 |
0,23 |
0,30 |
|
Содержание капролактама, мкг |
0 |
50 |
75 |
100 |
150 |
200 |
Контрольный фильтр и фильтры с нанесенными стандартными растворами помещают в стакан и обрабатывают аналогично пробам. Одновременно во все пробирки шкалы и пробы добавляют по 0,1 мл раствора 2 н. соляной кислоты и по 0,5 мл 10% раствора хлорного железа. Через 10 мин. сравнивают интенсивность окраски проб со шкалой стандартов. Окраска шкалы стандартов от зеленовато-желтой до красновато-коричневой, устойчива в течение 30 мин.
По шкале стандартов (см. табл. 98) колориметрируют пробы, отобранные на фильтры АФА-Х-11, определение же количества капролактама, поглощенного в воду в поглотителях Гернет, производят по другой стандартной шкале (табл. 99), приготовленной из стандартного раствора, полученного также после гидролиза капролактама.
Таблица 99
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
|
Раствор гидроксамовой кислоты, полученной после гидролиза капролактама, мл |
0 |
0,08 |
0,12 |
0,15 |
0,23 |
0,30 |
|
Контрольный раствор, мл |
3 |
2,92 |
2,88 |
2,85 |
2,77 |
2,70 |
|
Содержание капролактама, мкг |
0 |
50 |
75 |
100 |
150 |
200 |
Одновременно во все пробирки шкалы и пробы добавляют по 0,1 мл 2 н. раствора соляной кислоты, 0,5 мл 10% раствора хлорного железа, перемешивают и через 10 мин. колориметрируют.
Расчет см. выше.
Примечание. Количество капролактама, обнаруженное в смыве с фильтра и в пробе из поглотителя Гернет, суммируют.
ОКИСЬ ЭТИЛЕНА
|
|
Мол. вес 44,03 |
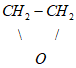
Бесцветный газ, при низких температурах — жидкость с эфирным запахом; удельный вес 0,8713 (20 °C), температура кипения 10,7 °C, упругость пара 1,095 мм рт. ст. (20 °C). Растворим в воде, эфире и спирте. Окись этилена способна к реакции со многими органическими и неорганическими веществами, полимеризируется, при нагревании с водой образует этиленгликоль, при 400 °C получается изомерный ацетальдегид.
Обладает наркотическим действием и сильным общетоксическим действием на организм.
Предельно допустимые концентрации окиси этилена: максимальная разовая и среднесуточная 0,03 мг/м3.
ОПРЕДЕЛЕНИЕ ОКИСИ ЭТИЛЕНА ПО ФОРМАЛЬДЕГИДУ [3]
Принцип определения
При поглощении окиси этилена водным раствором серной кислоты образуется этиленгликоль, последний окисляют йодной кислотой до формальдегида. Формальдегид определяют колориметрически с хромотроповой кислотой. Чувствительность определения 0,5 мкг в анализируемом объеме раствора. Определению мешают формальдегид, этиленгликоль.
Реактивы
1. Поглотительный раствор: 40% раствор серной кислоты.
2. Стандартный раствор окиси этилена. Готовят исходный раствор, для чего берут мерную колбу емкостью 50 мл, наливают в нее 10 — 15 мл 40% раствора серной кислоты, взвешивают, добавляют в колбу 2 — 3 капли окиси этилена и снова взвешивают. Раствор в колбе доводят до метки 40% раствором серной кислоты и тщательно перемешивают.
Рассчитывают содержание окиси этилена в 1 мл раствора и готовят рабочие стандартные растворы А и Б с содержанием 100 мкг/мл (раствор А) и 10 мкг/мл (раствор Б).
Стандартные растворы можно готовить из этиленгликоля.
Раствор А должен содержать 140 мкг/мл этиленгликоля, рабочий стандартный раствор — 14 мкг/мл, что соответствует 100 и 10 мкг окиси этилена.
3. Перйодат калия (KIO4), 3% раствор в 10% растворе серной кислоты. Растворение проводят при нагревании.
4. Сульфит натрия, 50% раствор. Раствор устойчив в течение 2 — 3 сут. Раствор готовят при нагревании.
5. Хромотроповая кислота. 0,15 г реактива растворяют в 5 мл воды, затем прибавляют 125 мл концентрированной серной кислоты. Раствор устойчив в течение 2 — 3 дней.
6. Серная кислота, концентрированная, 10 и 40% растворы.
Отбор пробы
а) Для определения разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 4 мл поглотительного раствора.
Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,25 — 0,3 л/мин. протягивают через два поглотительных прибора с пористой пластинкой N 1, содержащих по 4 мл поглотительного раствора, отмечая уровень жидкости. Отбор проводят непрерывно в течение суток, периодически добавляя воду до первоначального уровня. Допустимо проводить отбор 6 — 12 раз с перерывом в 2 — 4 ч в одни и те же поглотительные приборы.
Ход анализа
В колориметрическую пробирку из каждого поглотительного прибора вносят 3 мл пробы, приливают по 0,5 мл 3% раствора перйодата калия, перемешивают и оставляют на 30 мин. Далее добавляют по 3 капли раствора сульфита натрия для восстановления избытка перйодата калия, по 1,5 мл хромотроповой кислоты, перемешивают и погружают на 30 мин. в кипящую водяную баню. По охлаждении добавляют 2 мл воды и перемешивают. При этом коричневый фон, обусловленный выделением йода, исчезает и остается фиолетовое окрашивание, характерное для реакции формальдегида с хромотроповой кислотой. После охлаждения растворы переливают в кюветы с толщиной слоя 1 см и фотометрируют при длине волны 574 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание окиси этилена в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов (табл. 100).
Таблица 100
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор Б, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
3 |
2,95 |
2,9 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
1,0 |
|
Содержание окиси этилена, мкг |
0 |
0,5 |
1,0 |
2,0 |
4,0 |
6,0 |
8,0 |
10,0 |
20,0 |
Все пробирки шкалы обрабатывают аналогично пробам, фотометрируют и строят калибровочный график.
Шкалой стандартов можно пользоваться и для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Примечание. Очистку хромотроповой кислоты см. выше.
Расчет см. выше.
ТОЛУИЛЕНДИИЗОЦИАНАТ
|
(CH3)C6H3(NCO)2 |
Мол. вес 174,0 |
Прозрачная слегка желтоватая жидкость с неприятным запахом. Толуилендиизоцианат известен в двух изомерах: 2,4- и 2,6-толуилендиизоцианат. Перегоняется под вакуумом, в воде гидролизуется, хорошо растворяется в органических растворителях. Смесь изомеров 2,4- и 2,6-толуилендиизоцианатов в отношении 65:35 обладает температурой кипения 162 °C при 5 мм рт. ст.
Толуилендиизоцианат действует раздражающе на слизистые оболочки верхних дыхательных путей и глаз.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3 и среднесуточная 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ ТОЛУИЛЕНДИИЗОЦИАНАТА С ![]() -НАФТОЛОМ [8]
-НАФТОЛОМ [8]
Принцип определения
При взаимодействии диамина, образующегося при омылении толуилендиизоционата щелочью, с нитритом натрия в солянокислой среде происходит диазотирование. При сочетании диазосоединения с ![]() -нафтолом образуется соединение, окрашивающее раствор в оранжево-красный цвет.
-нафтолом образуется соединение, окрашивающее раствор в оранжево-красный цвет.
Чувствительность определения — 0,5 мкг в анализируемом объеме раствора.
Толуилендиамин, мешающий определению, задерживают на фильтре АФА.
Реактивы
1. Поглотительный раствор — 0,1 н. раствор едкого натра.
2. Стандартный раствор толуилендиизоцианата. Готовят исходный раствор в мерной колбе емкостью 50 мл, в которую наливают 15 — 20 мл ацетона, колбу взвешивают, в нее вносят 2 — 3 капли толуилендиизоцианата и снова взвешивают. Раствор в колбе доводят до метки ацетоном и хорошо перемешивают. По разности двух взвешиваний устанавливают количество толуилендиизоцианата в колбе и высчитывают его содержание в 1 мл раствора.
Рабочий стандартный раствор с содержанием 10 мкг/мл получают в мерной колбе емкостью 50 мл, в нее вносят такое количество исходного раствора, в котором содержится 500 мкг толуилендиизоцианата. Раствор в колбе доводят до метки поглотительным раствором.
3. Нитрит-бромидный реактив. 0,5 г бромистого натрия растворяют в 100 мл 0,3% раствора нитрита натрия.
4. ![]() -Нафтол, 0,1% раствор в 1 н. растворе едкого натра.
-Нафтол, 0,1% раствор в 1 н. растворе едкого натра.
Готовят за 10 мин. до применения.
5. Натр едкий, 1 н. раствор.
6. Соляная кислота, 0,2 н. раствор.
7. Нитрит натрия, 0,3% раствор.
Отбор пробы
а) Для определения максимальной разовой концентрации воздух со скоростью 0,5 л/мин. протягивают через поглотительный прибор с пористым фильтром N 1, наполненный 5 мл поглотительного раствора. Продолжительность отбора 20 — 30 мин.
б) Для определения среднесуточной концентрации воздух со скоростью 0,3 л/мин. протягивают через поглотительный прибор, наполненный 5 мл поглотительного раствора. Если в процессе отбора пробы происходит изменение объема, то периодически в поглотительный прибор вносят воду и доводят объем до первоначального уровня. Допустимо отбор пробы проводить 6 — 12 раз с перерывами в 2 — 4 ч в один и тот же поглотительный прибор.
Ход анализа
В пробирку переносят 2,5 мл пробы, добавляют 2,8 мл 0,2 н. раствора соляной кислоты. Пробу помещают в сосуд со льдом на 5 мин. После этого в пробирку вносят 0,2 мл нитрит-бромидного реактива, содержимое перемешивают и добавляют 0,2 мл ![]() -нафтола. Раствор тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см при длине волны 496 нм по сравнению с контролем.
-нафтола. Раствор тщательно перемешивают и через 10 мин. измеряют оптическую плотность в кювете с толщиной слоя 1 см при длине волны 496 нм по сравнению с контролем.
Контроль готовят одновременно и аналогично пробе.
Содержание толуилендиизоцианата в анализируемом объеме определяют по заранее построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 101).
Таблица 101
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
|
Поглотительный раствор, мл |
2,5 |
2,45 |
2,4 |
2,3 |
2,1 |
1,9 |
1,7 |
1,5 |
|
|
Содержание толуилендиизоцианата, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ТИОФЕН
(Тиофуран)
|
|
Мол. вес 84,14 |
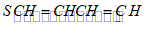
Бесцветная жидкость с температурой кипения 84 °C, температурой плавления — 38,3 °C. Плотность 1,064 (20,1 °C). Тиофен не растворим в воде, растворим в спирте, эфире, бензоле; устойчив к действию окислителей, легко реагирует с ртутью, дает металлоорганические соединения.
Пары тиофена вызывают раздражение слизистых оболочек дыхательных путей. Обладает наркотическим действием.
Предельно допустимая максимальная разовая концентрация тиофена 0,6 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ТИОФЕНА [1]
Принцип определения
Метод основан на измерении оптической плотности спиртового раствора тиофена при длине волны 231 нм.
Чувствительность определения — 2,5 мкг в анализируемом объеме раствора. Определению мешают вещества, поглощающие при длине волны 231 нм.
Реактивы
1. Поглотительный раствор — метиловый спирт, перегнанный при 64 — 65 °C.
2. Стандартные растворы тиофена. В мерную колбу емкостью 25 мл с притертой пробкой наливают 10 — 15 мл метилового спирта и взвешивают на аналитических весах, затем вносят 2 — 3 капли тиофена и снова взвешивают. Объем доводят метиловым спиртом до метки, тщательно перемешивают и по разности между первым и вторым взвешиванием рассчитывают содержание тиофена в 1 мл. Соответствующим разведением готовят стандартный раствор с содержанием 0,1 мг/мл, а из него разведением в 10 раз готовят рабочий стандартный раствор с содержанием 10 мкг/мл.
3. Тиофен, х.ч.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 0,1 — 0,2 л/мин. протягивают через 2 — 3 поглотительных прибора с пористой пластинкой N 1, содержащих 6 мл метилового спирта. Во время отбора пробы поглотительный прибор помещают в сосуд со льдом. Продолжительность отбора 20 — 30 мин.
Ход анализа
Содержимое каждого поглотительного прибора переливают в кварцевую кювету с толщиной слоя 1 см и фотометрируют на спектрофотометре при длине волны 231 нм. В качестве контроля применяют метиловый спирт. Содержание тиофена в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 102).
Таблица 102
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,25 |
0,5 |
1 |
2 |
3 |
4 |
5 |
|
Метиловый спирт, мл |
5 |
4,75 |
4,5 |
4,0 |
3,0 |
2,0 |
1,0 |
0 |
|
Содержание тиофена, мкг |
0 |
2,5 |
5,0 |
10,0 |
20,0 |
30,0 |
40,0 |
50,0 |
Растворы шкалы стандартов фотометрируют в тех же условиях, что и пробы.
Расчет см. выше.
ДИВИНИЛ
(1,3-Бутадиен)
|
CH2=CH-CH=CH2 |
Мол. вес 54,09 |
Газ с неприятным запахом, не растворимый в воде и растворимый в органических растворителях. Температура кипения 4,5 °C, температура плавления -109 °C, легко полимеризируется.
Дивинил оказывает раздражающее действие на слизистые оболочки верхних дыхательных путей, глаз; может вызывать дерматиты, в высоких концентрациях действует как наркотик.
Предельно допустимые концентрации дивинила: максимальная разовая 3 мг/м3, среднесуточная 1 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ
ДИВИНИЛА В ПРИСУТСТВИИ ![]() -МЕТИЛСТИРОЛА
-МЕТИЛСТИРОЛА
И ИЗОПРОПИЛБЕНЗОЛА [4]
Принцип определения
Количественное определение дивинила проводят по спектрам поглощения изооктанового раствора трехкомпонентной системы дивинила, изопропилбензола и альфа-метилстирола при длинах волн 224 и 245 нм.
Чувствительность определения — 0,1 мкг/мл раствора. Определению мешают вещества, поглощающие в рабочих длинах волн 224 и 245 нм.
Реактивы
1. Силикагель марки КСМ и МСМ с зернением 1 мм, предварительно обработанный.
2. Изооктан эталонный, оптически чистый. Для получения оптически чистого изооктана эталонный изооктан дополнительно очищается с помощью колонки, заполненной силикагелем, до получения растворителя с оптической плотностью ~ 0,175 относительно дистиллированной воды при 220 нм в кювете с толщиной слоя 1 см.
3. Стеклянная вата.
Отбор пробы
Для определения разовой концентрации воздух со скоростью 1 л/мин. протягивают через V-образную стеклянную трубку диаметром 4 мм, заполненную 0,5 г силикагеля. Трубка закрыта тампонами из стеклянной ваты и стеклянными заглушками. При отборе пробы заглушки снимают. При наличии запаха дивинила достаточно отобрать 1 л воздуха, при отсутствии запаха следует отобрать 5 — 10 л воздуха.
Ход анализа
Силикагель из V-образной трубки переносят в пробирку с притертой пробкой, заливают 4 мл изооктана, тщательно перемешивают, закрывают пробкой и оставляют в горизонтальном положении при частом встряхивании на 1 ч. Прозрачную жидкость над слоем силикагеля сливают, помещают в кварцевую кювету с толщиной слоя 1 см и измеряют оптическую плотность на спектрофотометре. Как эталон сравнения ставят изооктан, полученный путем обработки чистого силикагеля.
При длине волны 224 нм находят суммарную оптическую плотность (E1), при длине волны 245 нм находят оптическую плотность (E2) мешающей примеси.
Расчет анализа
Концентрацию дивинила (C) в микрограммах на 1 мл вычисляют по формуле:
C = 2,5 (E — 0,47 E2).
Концентрацию дивинила в миллиграммах на 1 м3 воздуха рассчитывают по обычной формуле (см. выше).
Примечание. Коэффициенты 0,47 и 2,5 постоянные. 0,47 — постоянное соотношение оптической плотности ![]() -метилстирола при 224 и 245 нм. Оптическая плотность дивинила с концентрацией 1 мкг/мл при 224 нм составляет 1 / 4 = 2,5.
-метилстирола при 224 и 245 нм. Оптическая плотность дивинила с концентрацией 1 мкг/мл при 224 нм составляет 1 / 4 = 2,5.
ОПРЕДЕЛЕНИЕ ОБЩЕГО КОЛИЧЕСТВА ОЗОНА И ОКСИДАНТОВ
С РОДАНИСТЫМ АММОНИЕМ [2]
Принцип определения
Метод основан на реакции взаимодействия оксидантов с солью Мора в кислой среде с образованием ионов трехвалентного железа, которое определяется колориметрически в виде железороданидного комплекса.
Чувствительность определения по озону — 0,3 мкг в анализируемом объеме раствора. Определению не мешают сероводород, формальдегид, акролеин, углеводороды. Определению мешает сернистый газ в концентрациях, превышающих оксиданты в 50 — 100 раз. Двуокись азота входит в состав оксидантов.
Реактивы
1. Поглотительный раствор. 0,1 г соли Мора растворяют в 100 мл дистиллированной воды, добавляют 10 мл 1 М азотной кислоты и 10 мл ацетона. Поглотительный раствор должен быть свежеприготовленный и стоять на холоду.
2. Стандартный раствор перекиси водорода. Для приготовления стандартного раствора 5 — 10 мл 30% раствора перекиси водорода растворяют в 500 мл дистиллированной воды в мерной колбе. Для титрования в коническую колбу берут 10 мл раствора, прибавляют 200 мл воды, 20 мл серной кислоты (1:3) и титруют 0,1 н. раствором перманганата калия до появления розового окрашивания. 1 мл 0,1 н. раствора перманганата калия соответствует 1,7 мг перекиси водорода. Путем соответствующего разбавления готовят раствор с содержанием 10 мкг/мл и рабочий стандартный раствор с содержанием 1 мкг/мл. Стандартные растворы должны быть свежеприготовленными.
3. Азотная кислота, 1 М раствор. Перед приготовлением 1 М раствора концентрированную азотную кислоту продувают током воздуха с целью удаления окислов азота.
4. Соль Мора FeSO4·(NH4)2SO4·6H2O, перекристаллизованная.
5. Ацетон.
6. Роданистый аммоний. Реактив должен быть бесцветным или слегка розоватым.
7. Роданистый аммоний, 30% раствор. Раствор должен быть бесцветным или слегка розоватым. При хранении на холоду устойчив в течение нескольких дней.
8. Серная кислота 1:3 (по объему).
9. Перманганат калия, 0,1 н. раствор.
Отбор пробы
Исследуемый воздух со скоростью 0,5 л/мин. протягивают через два поглотительных прибора, наполненных по 5 мл поглотительного раствора.
Ход анализа
Из каждого поглотительного прибора переносят 5 мл пробы в колориметрические пробирки и приливают по 2 мл 30% раствора роданистого аммония. Через 5 — 10 мин. фотометрируют образующуюся окраску в кюветах с толщиной слоя 1 см при длине волны 480 — 490 нм по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание оксидантов в анализируемом объеме раствора определяют по предварительно построенному градуировочному графику. Для построения градуировочного графика готовят шкалу стандартов (табл. 103).
Таблица 103
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
|
Рабочий стандартный раствор, мл |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Поглотительный раствор, мл |
5 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
3,0 |
|
Содержание перекиси водорода, мкг |
0 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
2,0 |
|
Эквивалентное содержание озона, мкг |
0 |
0,28 |
0,57 |
0,85 |
1,13 |
1,41 |
2,82 |
Шкалу стандартов обрабатывают аналогично пробе, измеряют оптическую плотность растворов и строят график. Шкалу стандартов можно использовать для визуального определения. В этом случае ее строят одновременно с пробами. Окраска устойчива в течение 30 — 40 мин.
Расчет анализа см. выше.
Примечание. Поглотительные приборы с пористой пластинкой или поглотительный прибор Зайцева перед отбором пробы воздуха должны быть отмыты от следов железа. Для этого предварительно хорошо вымытые поглотительные приборы заливают поглотительным раствором на 4 — 5 ч; раствор выливают, поглотители промывают еще 2 — 3 раза поглотительным раствором с помощью резинового баллона, раствор сливают, добавляют 2 мл роданистого аммония и измеряют его оптическую плотность. Оптическая плотность раствора должна быть меньше 0,01. Отмытые поглотительные приборы хранят залитыми дистиллированной водой и закрытыми заглушками.
ИНГИБИТОРЫ АТМОСФЕРНОЙ КОРРОЗИИ
Ингибиторами называются вещества, тормозящие химические реакции. Они защищают от атмосферной коррозии черные и цветные металлы.
НИТРИТ ДИЦИКЛОГЕКСИЛАМИНА (НДА)
|
|
Мол. вес 228,1 |
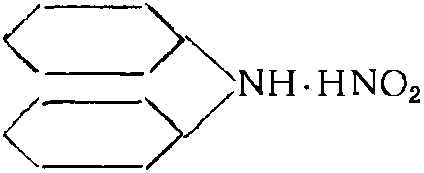
Твердое вещество белого цвета. Температура плавления 180 °C, упругость пара 0,1 · 103 мм рт. ст. Плохо растворимо в воде, растворимо в этаноле, метаноле, ацетоне. Токсично. Предельно допустимая концентрация: максимальная разовая 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ НИТРИТА ДИЦИКЛОГЕКСИЛАМИНА (НДА)
С РЕАКТИВОМ ГРИССА-ИЛОСВАЯ [9]
Принцип определения
При взаимодействии НДА с реактивом Грисса-Илосвая образуется азокраситель, окрашивающий раствор в розовый цвет.
Чувствительность определения — 0,5 мкг НДА в анализируемом объеме раствора. Определению мешает двуокись азота.
Реактивы
1. Поглотительный раствор — дистиллированная вода.
2. Стандартный раствор НДА. Исходный стандартный раствор с содержанием 100 мкг/мл готовят растворением 0,01 г НДА в 100 мл дистиллированной воды. Рабочий стандартный раствор с содержанием 10,0 мкг/мл готовят соответствующим разведением исходного раствора.
Остальные реактивы — см. «Определение двуокиси азота».
Отбор пробы
Для определения разовой концентрации воздух со скоростью 2 — 3 л/мин. протягивают через фильтр АФА, помещенный в патрон, соединенный с двумя поглотительными приборами Рыхтера, наполненными по 6 мл воды.
Ход анализа
В пробирки вносят по 5 мл пробы из каждого поглотительного прибора. Фильтр извлекают из патрона, переносят в стакан и заливают 3 мл воды, отжимают на стенке стакана стеклянной палочкой и снова обрабатывают 3 мл воды. В пробирку помещают 5 мл раствора. Во все пробирки добавляют по 0,5 мл реактива Грисса-Илосвая, перемешивают, оставляют стоять 20 — 30 мин. и фотометрируют при длине волны 530 нм в кювете с толщиной слоя 1 см по сравнению с контролем, который готовят одновременно и аналогично пробе. Содержание НДА в анализируемом объеме раствора определяют по предварительно построенному калибровочному графику. Для построения графика готовят шкалу стандартов (табл. 104).
Таблица 104
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Поглотительный раствор, мл |
5 |
4,95 |
4,9 |
4,8 |
4,6 |
4,4 |
4,2 |
4,0 |
|
Содержание НДА, мкг |
0 |
0,5 |
1 |
2 |
4 |
6 |
8 |
10 |
Все пробирки шкалы обрабатывают аналогично пробе, измеряют оптическую плотность и строят график.
Расчет см. выше.
МЕТАНИТРОБЕНЗОАТГЕКСАМЕТИЛЕНИМИН (Г-2) [9]
|
|
Мол. вес 226,0 |
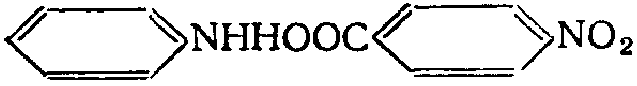
Твердое вещество белого цвета. Температура плавления 133 °C, упругость пара 1 · 106 мм рт. ст. Плохо растворим в воде, растворим в этаноле. Относится к токсическим соединениям.
Предельно допустимая концентрация: максимальная разовая 0,02 мг/м3.
ОПРЕДЕЛЕНИЕ МЕТАНИТРОБЕНЗОАТАГЕКСАМЕТИЛЕНИМИНА
(ИНГИБИТОРА — Г-2) С n-ДИМЕТИЛАМИНОБЕНЗАЛЬДЕГИДОМ [9]
Принцип определения
Метод основан на восстановлении метанитробензотагексаметиленимина с последующим определением по реакции конденсации с парадеметиламинобензальдегидом.
Чувствительность определения — 3 мкг в анализируемом объеме раствора.
Определению мешают некоторые первичные ароматические амины.
Реактивы
1. Стандартные растворы Г-2.
Стандартный раствор Г-2 готовят растворением 0,01 г ингибитора в 100 мл дистиллированной воды. Содержание Г-2 в растворе 100 мкг/мл.
2. Парадиметиламинобензальдегид, 5% раствор в 5% растворе соляной кислоты.
3. Соляная кислота, концентрированная.
4. Соляная кислота, 5% раствор.
5. Гранулированный цинк.
Отбор пробы
Для определения разовой концентрации исследуемый воздух со скоростью 3 л/мин. протягивают через фильтр АФА-В, помещенный в патрон, соединенный с двумя поглотительными приборами Рыхтера, наполненными по 6 мл воды. Продолжительность отбора 20 — 30 мин.
Ход анализа
В пробирки вносят 3 мл пробы из каждого поглотительного прибора, а фильтр извлекают и переносят в стакан. Фильтр смачивают 1 — 2 каплями этилового спирта, заливают 3 мл воды, промывают и отжимают стеклянной палочкой на воронке. Операцию повторяют.
В колориметрические пробирки переносят 3 мл жидкости. Во все пробы добавляют по 0,5 мл концентрированной соляной кислоты и по одной грануле цинка. Восстановление проводят при 100 — 110 °C на песчаной бане в течение 5 мин. После охлаждения пробы отфильтровывают в колориметрические пробирки, приливают 1,5 мл 5% раствора парадиметиламинобензальдегида в 5% растворе соляной кислоты, перемешивают, оставляют стоять в течение 10 мин. и фотометрируют в кювете с толщиной слоя 1 см при длине волны 445 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание Г-2 в анализируемом объеме определяют по предварительно построенному градуировочному графику, который строят по шкале стандартов (табл. 105).
Таблица 105
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
0 |
1 |
2 |
3 |
4 |
5 |
6 |
|
Стандартный раствор, мл |
0 |
0,03 |
0,05 |
0,1 |
0,2 |
0,4 |
0,8 |
|
Поглотительный раствор, мл |
3 |
2,97 |
2,95 |
2,9 |
2,8 |
2,6 |
2,2 |
|
Содержание Г-2, мкг |
0 |
3 |
5 |
10 |
20 |
40 |
80 |
Шкалу стандартов обрабатывают аналогично пробам и строят график. Определение можно проводить и визуально.
Расчет анализа см. выше.
МАСЛОРАСТВОРИМАЯ СОЛЬ ДИЦИКЛОГЕКСИЛАМИНА (МСДА)
(C6H11)2HChH2h-1COOH
Маслорастворимая соль дициклогексиламина представляет собой бурое, пастообразное вещество с температурой плавления 350 °C, не растворимое в воде, растворимое в спирте (3 г на 100 г), ацетоне и бензоле. Обладает токсическим действием. Предельно допустимая концентрация: максимальная разовая 0,008 мг/м3.
ОПРЕДЕЛЕНИЕ МАСЛОРАСТВОРИМОЙ СОЛИ ДИЦИКЛОГЕКСИЛАМИНА (МСДА)
С ДИЭТИЛДИТИОКАРБАМАТОМ НАТРИЯ [9]
Принцип определения
Метод основан на образовании медных солей жирных кислот с последующим определением их по реакции с диэтилдитиокарбаматом натрия.
Чувствительность определения — 5 мкг в анализируемом объеме раствора. Определению мешают жирные кислоты.
Реактивы
1. МСДА.
2. Стандартные растворы МСДА в хлороформе.
Для приготовления исходного стандартного раствора с содержанием 1 мг/мл в бюксе взвешивают 25 мг. МСДА растворяют в хлороформе, переносят в мерную колбу емкостью 25 мл и доливают хлороформом до метки.
Из исходного стандартного раствора путем соответствующего разбавления готовят рабочий стандартный раствор, содержащий 100 мкг/мл.
3. Хлороформ, х.ч.
4. Триэтаноламин, х.ч. 1 М раствор.
5. Уксусная кислота, ледяная.
6. Уксусная кислота, 1 М раствор.
7. Медь сернокислая, 6,5% водно-спиртовой раствор.
8. Составной раствор готовят из 9 частей 1 М триэтаноламина, 1 части 1 М уксусной кислоты и 10 частей 6,5% раствора сернокислой меди (раствор применяется свежеприготовленный).
9. Бутиловый спирт.
10. Диэтилдитиокарбамат натрия, 0,1% раствор в бутиловом спирте (раствор должен быть свежеприготовленный).
11. Беззольные фильтры «синяя лента».
Отбор пробы
Воздух со скоростью 3 — 10 л/мин. протягивают через бумажный фильтр, помещенный в патрон.
Ход анализа
Фильтр с отобранной пробой помещают в коническую колбу, заливают 6 мл хлороформа. Фильтр промывают в хлороформе, отжимают стеклянной палочкой на воронке и добавляют 5 мл составного раствора. Колбу помещают в аппарат для встряхивания на 1 1/2 — 2 мин.
Содержимое колбы переносят в делительную воронку, разделяют смесь и сливают хлороформный слой через воронку с беззольным фильтром, предварительно смоченным хлороформом, в пробирку с притертой пробкой. К 5 мл хлороформного экстракта добавляют 0,5 мл 0,1% раствора диэтилдитиокарбамата натрия в бутиловом спирте, перемешивают и фотометрируют в кювете с толщиной слоя 1 см при длине волны 440 нм по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание МСДА в анализируемом объеме определяют по предварительно построенному калибровочному графику.
Для построения калибровочного графика готовят шкалу стандартов, при этом рабочий стандартный раствор наносят на фильтры (табл. 106).
Таблица 106
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
|
Содержание МСДА, мкг |
0 |
5 |
10 |
20 |
40 |
60 |
80 |
100 |
Одновременно готовят контрольный фильтр. Контрольный фильтр и фильтры, содержащие различное количество МСДА, помещают в колбы с притертой пробкой и далее обрабатывают аналогично пробе, измеряют оптическую плотность растворов и строят график.
Расчет см. выше.
1,4-НАФТОХИНОН
|
|
Мол. вес 158,15 |
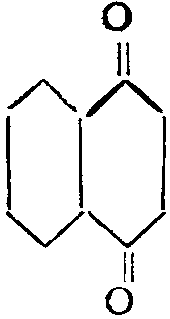
Ярко-желтые кристаллы с температурой плавления 125°, легко возгоняющиеся. 1,4-Нафтохинон не растворяется в воде, растворяется в органических растворителях, окислитель.
Применяется в производстве синтетических красителей. Токсичен, обладает резко выраженным раздражающим свойством, действует на нервную систему, почки.
2,3-ДИХЛОР-1,4-НАФТОХИНОН
|
|
Мол. вес 227,0 |
Твердое кристаллическое вещество желтого цвета с температурой плавления 193 °C, растворимое в горячем спирте, не растворимое в воде. Применяется в химической промышленности. Токсичен, обладает резорбтивным и местным раздражающим действием на теплокровных животных.
Предельно допустимые концентрации: максимальная разовая 0,05 мг/м3, среднесуточная 0,05 мг/м3.
СПЕКТРОФОТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ 1,4-НАФТОХИНОНА
И 2,3-ДИХЛОР-1,4-НАФТОХИНОНА [10]
Принцип определения
Метод основан на способности 2,3-дихлор-1,4-нафтохинона поглощать свет при длине волны 280 нм, тогда как 1,4-нафтохинон в этой области прозрачен. В то же время эти соединения имеют интенсивную полосу поглощения при длине 246 нм. Использование метода Фирордта позволяет по величине оптической плотности смеси двух компонентов определять концентрацию каждого из них.
Чувствительность определения — 1 мкг/мл.
Реактивы
1. Этанол.
2. Фильтры АФА-ХА-18.
Отбор пробы
Воздух протягивают со скоростью 2 л/мин. через фильтр АФА, закрепленный в патроне, последовательно соединенный с двумя поглотительными приборами, содержащими по 10 мл этанола.
Отбор проб проводят при охлаждении.
Ход анализа
Фильтр переносят в стакан и вливают 10 мл этанола. В течение 10 мин. помешивают стеклянной палочкой. Фильтр осторожно отжимают и промывают его 5 мл этанола. Экстракты соединяют, сюда же переносят и содержимое поглотительных приборов. Измеряют объем пробы.
Раствор (4 мл) переносят в кювету с толщиной слоя 1 см и измеряют оптические плотности при 246 и 280 нм. Измерения проводят по отношению к этанолу.
Предварительно из стандартных этанольных растворов, в которых содержится по 100 мкг/мл 1,4-нафтохинона или 2,3-дихлор-1,4-нафтохинона, готовят стандартную шкалу. В каждой пробирке шкалы должно содержаться по 1 — 3 — 5 — 7 — 10 мкг/мл раствора того или другого вещества. Измеряют оптические плотности растворов 2,3-дихлор-1,4-нафтохинона и отдельно 1,4-нафтохинона при 246 нм.
Кроме того, измеряют оптические плотности растворов 2,3-дихлор-1,4-нафтохинона при длине волны 280 нм.
Строят три градуировочных графика:
1) для 2,3-дихлор-1,4-нафтохинона при 246 нм,
2) для 1,4-нафтохинона при 246 нм,
3) для 2,3-дихлор-1,4-нафтохинона при 280 нм.
По градуировочному графику (3) находят концентрацию 2,3-дихлор-1,4-нафтохинона на основании полученной величины оптической плотности при измерении пробы.
Затем по градуировочному графику (1) находят величину оптической плотности, соответствующую найденной концентрации 2,3-дихлор-1,4-нафтохинона. Разность оптических плотностей указывает на величину оптической плотности, соответствующую 1,4-нафтохинону. Концентрацию 1,4-нафтохинона определяют по калибровочному графику (2). Таким образом находят концентрации того и другого соединения в 1 мл исследуемого раствора. При расчете учитывают объем всей пробы.
Расчет см. выше.
БЕНЗ(А)ПИРЕН <1>
———————————
<1> По старой номенклатуре — 3,4-бензпирен
|
|
Мол. вес 252,3 |
Бенз(а)пирен (БП) твердое кристаллическое вещество, температура плавления 179 °C, стойкое к внешним воздействиям.
БП относится к пятикольчатым ароматическим углеводородам, обладает высокой канцерогенной активностью.
Источниками образования и выделения БП в атмосферный воздух и другие внешние среды являются процессы термической переработки органического сырья, сжигания топлив и др.
Среднесуточная предельно допустимая концентрация 0,1 мкг на 100 м3 воздуха. При изучении уровня загрязнения атмосферного воздуха бенз(а)пиреном применяют аспирационный и седиментационный способы отбора проб. При аспирационном способе исследуемый воздух протягивают через фильтры ФПП-15, ФПА-15 со скоростью до 250 м3/ч в течение от 20 мин. до нескольких часов. Обычно накапливают до 4 г пыли, в которой определяют количество БП. В ряде случаев отбор проб проводят в поглотительные приборы, наполненные бензолом, со скоростью 0,5 — 1 л/мин. в течение 20 — 50 мин.
При седиментационном способе отбирают снеговые пробы или собирают пыль в специальные сосуды.
Из анализируемой пробы извлекают фракции, содержащие БП, очищают их от сопутствующих примесей хроматографированием.
Количественное определение БП проводят методом квазилинейчатых спектров люминесценции (эффект Шпольского), позволяющим устанавливать БП в концентрациях 10-8 — 10-10 г/мл (0,01 — 0,0001 мкг/мл).
Подготовка проб к анализу, специальная обработка посуды, растворителей, а также проведение анализа подробно изложены в двух нормативных документах, утвержденных Министерством здравоохранения СССР в 1972 г. [5, 6].
ЛИТЕРАТУРА
1. Аигина Е.Л., Лопухова А.И. и др. Определение тиофена в воздухе спектрофотометрическим методом. Гиг. и сан., 1966, 6.
2. Дмитриев М.Т., Соловьева Т.В. и др. Определение оксидантов. Гиг. и сан., 1972, 2.
3. Крылова Н.А. Определение окиси этилена в атмосферном воздухе, Гиг. и сан., 1961, 10.
4. Манита М.Д. Спектрофотометрический метод определения дивинила в присутствии альфа-метилстирола и изопропилбензола. Гиг. и сан., 1965, 8.
5. Методические указания по отбору проб из объектов внешней среды и их подготовке к анализу на канцерогенные полициклические ароматические углеводороды Министерства здравоохранения СССР. М., 1972, N 930-71.
6. Методические указания по определению канцерогенных полициклических ароматических углеводородов, в частности, бенз(а)пирена, в различных промышленных и природных продуктах Министерства здравоохранения СССР. М., 1972, N 931-71.
7. Русских А.А. Колориметрический метод определения капролактама в воздухе. Гиг. труда, 1965, 9.
8. Семенов В.А., Нехорошева Е.В. и др. Модификация колориметрического метода 2,4-толуилендиизоцианата с ![]() -нафтолом. Гиг. и сан., 1971, 4.
-нафтолом. Гиг. и сан., 1971, 4.
9. Соловьева Т.В., Евсеенко Н.С. и др. Определение ингибиторов атмосферной коррозии. Гиг. и сан., 1972, 1.
10. Черниченко Н.А. Спектрофотометрическое определение 1,4-нафтохинона в воздухе. Инструкция Киевского института общей и коммунальной гигиены им. А.Н.
Глава XV
САЖА, ПЫЛЬ, ДВУОКИСЬ КРЕМНИЯ В ПЫЛИ
САЖА
Сажа — продукт неполного сгорания или термического разложения углеродистых веществ. В различных видах сажи содержится углерода от 88,5 до 99,9%, водорода от 0,1 до 1%, кислорода от 0,01 до 10% и минеральных веществ от 0,01 до 0,6%. Сажа — высокодисперсный порошок, размер частиц ее от 10 до 600 ммк, удельный вес 1,6 — 2 г/см3. Сажа не растворяется ни в кислотах, ни в щелочах. Сажа применяется в полиграфической, лакокрасочной, резиновой промышленности. В производстве сажи, при большой запыленности, отмечены заболевания верхних дыхательных путей, органов дыхания и пищеварения, наблюдаются раздражения роговицы глаз и конъюнктивы. Некоторые виды сажи (например, полученные из пека) могут вызывать образование на коже злокачественных опухолей. Увеличение заболеваемости раком легких иногда объясняют наличием в воздухе сажи, в которой найдено до 0,03% 3,4-бензпирена. Сажа значительно уменьшает прозрачность атмосферы, что приводит к снижению освещенности, видимости и ультрафиолетовой радиации.
Предельно допустимые концентрации сажи (копоти): максимальная разовая 0,15 мг/м3, среднесуточная 0,05 мг/м3.
ОПРЕДЕЛЕНИЕ САЖИ. МЕТОД 1 [5]
Принцип определения
Сажу улавливают на бумажный фильтр. Степень почернения фильтра сопоставляют со шкалой, приготовленной из суспензии чистой сажи, нанесенной в определенных количествах на фильтровальную бумагу.
Чувствительность определения — 2 мкг. Мешают вещества, окрашивающие фильтр.
Аппаратура и реактивы
1. Патрон для отбора проб, внутренний диаметр 5 мм.
2. Беззольные фильтры («синяя лента»).
3. Вазелиновое масло.
4. Сапонин.
5. Стандартная шкала для определения сажи. Ее приготовляют следующим образом. Навеску сажи (около 120 мг), полученную путем сжигания чистого вазелинового масла, тщательно растирают в фарфоровой ступке с 2 — 3 мл дистиллированной воды, к которой прибавляют крупинку сапонина. После этого полученную смесь переносят в колбу емкостью 1 л, доводят водой до метки и тщательно взбалтывают. При этом получается равномерная и хорошо стабилизированная суспензия. При наличии на поверхности суспензии отдельных плавающих конгломератов сажи их осторожно снимают прикосновением фильтровальной бумаги.
Часть суспензии (500 мл) выпаривают в доведенной до постоянного веса фарфоровой чашке и снова высушивают до постоянного веса. Определяют истинную концентрацию сажи в стандартной суспензии. Она оказывается близкой к 0,1 мг/мл. Из этой основной суспензии разведением в мерной колбе приготовляют более слабую, с содержанием 10 мкг/мл.
Определенные объемы основной суспензии или ее десятикратного разведения фильтруют через бумажный фильтр, зажатый в описанном выше патроне. После фильтрования патрон промывают несколько раз водой, развинчивают, фильтр извлекают и высушивают при комнатной температуре. Фильтровальная бумага должна быть достаточно плотной и не пропускать частички сажи, что проверяют пробной фильтрацией суспензии через двойной фильтр. В этих условиях на втором листке фильтра не должно быть заметно почернения. Получающуюся серию фильтров наклеивают клейстером из рисового крахмала на ватманскую бумагу, покрывают тонким прозрачным стеклом и окантовывают по краям. Шкала может служить неопределенно долгое время.
Всего в шкале делается 10 ступеней: 2; 4; 6; 8; 10; 15; 20; 30; 40 и 50 мкг сажи.
Отбор пробы
а) Для определения разовой концентрации пробы отбирают с подветренной стороны от дымящего объекта. Воздух протягивают через бумажный фильтр, укрепленный в патроне, в течение 20 — 30 мин. со скоростью 1 л/мин.
б) Для определения среднесуточной концентрации воздух протягивают непрерывно в течение суток через такой же фильтр. Допустимо вместо круглосуточной аспирации отбирать пробу через один и тот же фильтр с перерывом в 2 — 4 ч. Общий объем протянутого воздуха должен составить 240 л; скорость протягивания воздуха 1 л/мин.
Ход анализа
Извлеченный из патрона фильтр сопоставляют по окраске со шкалой и подбирают наиболее близкую к окраске фильтра ступень шкалы.
Расчет анализа см. выше.
ОПРЕДЕЛЕНИЕ САЖИ. МЕТОД 2 [3]
Принцип определения
Сажу вместе с пылью улавливают на фильтре АФА-В. При обработке фильтра дихлорэтаном материал фильтра (перхлорвинил) и органические вещества пыли растворяются в дихлорэтане, основная высокодисперсная углеродистая составная часть сажи переходит во взвешенное суспендированное состояние, а минеральная часть пыли оседает на дно. Содержание сажи в суспензии определяют или визуально по почернению мембранного фильтра при помощи сопоставления со стандартной шкалой, или фотометрически.
Чувствительность определения — 2 мкг.
Методом можно определить визуально 0,5 мкг, фотометрически 3 мкг сажи.
Аппаратура и реактивы
1. Мембранные фильтры N 5.
2. Вазелиновое масло.
3. Дихлорэтан (хранить в темной склянке).
4. Сапонин.
5. Основная суспензия сажи.
Навеску сажи около 20 мг получают путем сжигания чистого вазелинового масла, тщательно растирают в фарфоровой ступке с 3 — 4 мл дихлорэтана, к которому прибавляют крупинку сапонина. Смесь переносят в колбу емкостью 100 мл, доводят дихлорэтаном до метки и тщательно взбалтывают. При этом получают равномерную и хорошо стабилизированную суспензию. Если на поверхности суспензии имеются отдельно плавающие конгломераты сажи, то их осторожно снимают прикосновением фильтровальной бумаги. В бюксе выпаривают 30 мл суспензии и доводят до постоянного веса. В основной суспензии сажи должно содержаться около 200 мкг/мл.
6. Стандартную суспензию сажи с содержанием 10 мкг/мл готовят из основной суспензии соответствующим разведением дихлорэтаном в мерной колбе.
Приготовление стандартной шкалы
Для приготовления стандартной шкалы определенные объемы стандартной суспензии сажи фильтруют через мембранные фильтры N 5, зажатые в патроне и колбе для отсасывания.
После фильтрования патрон каждый раз промывают дихлорэтаном и фильтры извлекают. Описанным способом приготовляют шкалу, отвечающую следующим количествам сажи: 0,5; 1; 2; 4; 7; 10; 15; 20 и 40 мкг. Серию фильтров наклеивают клейстером из рисового крахмала на бумагу и покрывают стеклом. Шкала устойчива длительное время.
Отбор пробы
Исследуемый воздух в количестве не менее 400 л протягивают через фильтр АФА-В-18, укрепленный в патроне, со скоростью 50 — 60 л/мин.
Ход анализа
а) Визуальное определение
Фильтр осторожно извлекают из патрона пинцетом и, если нужно, определяют содержание пыли. Опрессованные края фильтра обрезают и вкладывают в центрифужную пробирку с притертой пробкой. В пробирку вливают 3 мл дихлорэтана, закрывают пробкой и взбалтывают до полного растворения фильтра и получения однородной суспензии. При этом материал фильтра (перхлорвинил) и органические вещества пыли растворяются в дихлорэтане, основная высокодисперсная углеродистая составная часть сажи переходит во взвешенное суспендированное состояние, а минеральная часть пыли оседает на дно пробирки. Через 2 — 5 мин. суспензию из пробирки декантируют в пробирку с притертой пробкой и меткой 5 мл. В центрифужную пробирку вливают еще 2 мл дихлорэтана и также обрабатывают содержимое пробирки. Через 2 — 5 мин. остаточную суспензию декантируют в ту же пробирку. Из пробирки пипеткой берут 0,5 или 1 мл суспензии в зависимости от количества сажи и вносят на мембранный фильтр N 5, укрепленный в патроне и колбе для отсасывания, суспензию фильтруют. После фильтрования фильтр вынимают из патрона и содержание сажи определяют визуально, сопоставляя почернение фильтра со стандартной шкалой.
б) Фотометрическое определение
Фильтр обрабатывают в два приема 5 — 10 мл дихлорэтана, как указано выше (см. «Визуальное определение»). Через 2 — 5 мин. суспензию декантируют в пробирку с притертой пробкой. После перемешивания всю суспензию в количестве 5 мл выливают в кювету с толщиной слоя 1 см и фотометрируют с сине-фиолетовым светофильтром с максимумом пропускания при 400 нм. Количество сажи в пробе определяют по калибровочному графику.
Построение калибровочного графика
Для построения калибровочного графика в пробирки с меткой 5 мл помещают чистые фильтры АФА-В с обрезанными опрессованными краями. На фильтры наносят стандартную суспензию сажи в количествах 0,3; 0,6; 1; 1,5; 2; 3; 4 и 5 мл и доводят дихлорэтаном до объема 5 мл. Приготовленные растворы соответствуют 3; 6; 10; 15; 20; 30; 40 и 50 мкг сажи в 5 мл раствора. Содержимое пробирок взбалтывают до полного растворения фильтра, выливают в кюветы и фотометрируют. Найденные величины оптической плотности помещают в графике напротив величин содержания сажи (мкг) в анализируемом объеме. Контрольную пробу готовят из чистого фильтра АФА-В-18 с обрезанными опрессованными краями, растворенного в 5 мл дихлорэтана.
Расчет анализа см. выше.
ПЫЛЬ
Пыль относится к твердым атмосферным загрязнениям. Пыль, находящаяся в атмосферном воздухе, может быть как природного происхождения, так и результатом промышленной и бытовой деятельности человека. Концентрация взвешенных частиц является показателем загрязнения атмосферного воздуха.
В атмосферном воздухе населенных мест установлены предельно допустимые концентрации для пыли нетоксичной: максимальная разовая 0,5 мг/м3, среднесуточная 0,15 мг/м3.
ОПРЕДЕЛЕНИЕ ПЫЛИ [1]
Принцип определения
Воздух протягивают через фильтр из ткани ФПП-15-1,5. Концентрацию пыли определяют по привесу.
Чувствительность определения — 0,3 мг.
Аппаратура
Автомобильный аспиратор системы Института имени Ф.Ф. Эрисмана (рис. 28). Он состоит из Т-образной трубы (3), патрона-насадки (2), манометра (4) и разборного конусовидного стакана (1).
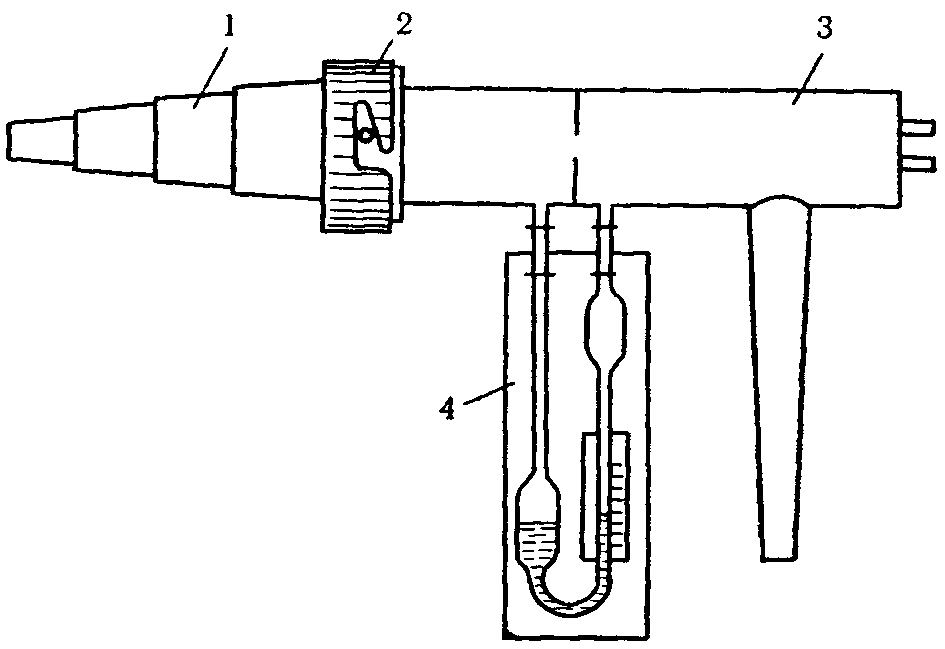
Рис. 28. Общий вид автомобильного аспиратора
а) Т-образная труба из разборного конусовидного стакана. Один конец трубы имеет втулку для укрепления патрона-насадки и два боковых отростка, к которым присоединяется манометр типа реометра, служащий для установления скорости отбора проб. Между этими отростками в трубе помещена диафрагма. Второй, противоположный, конец трубы имеет два отверстия, закрывающиеся дырчатой заслонкой, служащей для впуска воздуха в трубу мимо патрона насадки. Третий конец трубы (более узкий) служит для присоединения аспиратора с входным отверстием карбюратора автомашины.
б) Патрон-насадка (рис. 29), состоящий из корпуса, кассеты, крышки.
Корпус служит для крепления патрона в Т-образной трубке — аспирационной части автомобильного аспиратора. В корпус помещают кассету с фильтром, с наружной стороны его имеются два штифта, с помощью которых крепится крышка-насадка.
Кассета состоит из двух частей: собственно кассеты, дном которой является сетка, применяемая в аллонжах, и разрезного кольца, служащего для крепления фильтра. Кольцо по краям разреза снабжено двумя отверстиями диаметром по 2 мм, в которые вставляют пинцет для того, чтобы извлечь кольцо из кассеты. При обследовании рекомендуется иметь запас кассет (20 — 30) и в лабораторных условиях их заряжать взвешенными фильтрами.
Крышка служит для крепления кассеты с фильтром в патроне-насадке, а также для присоединения конусовидного стакана к патрону. С внутренней стороны крышка снабжена прокладкой для полноты герметичности крепления кассеты. На верхней стенке крышки имеется бортик, на который надевают разборный конусовидный стакан.
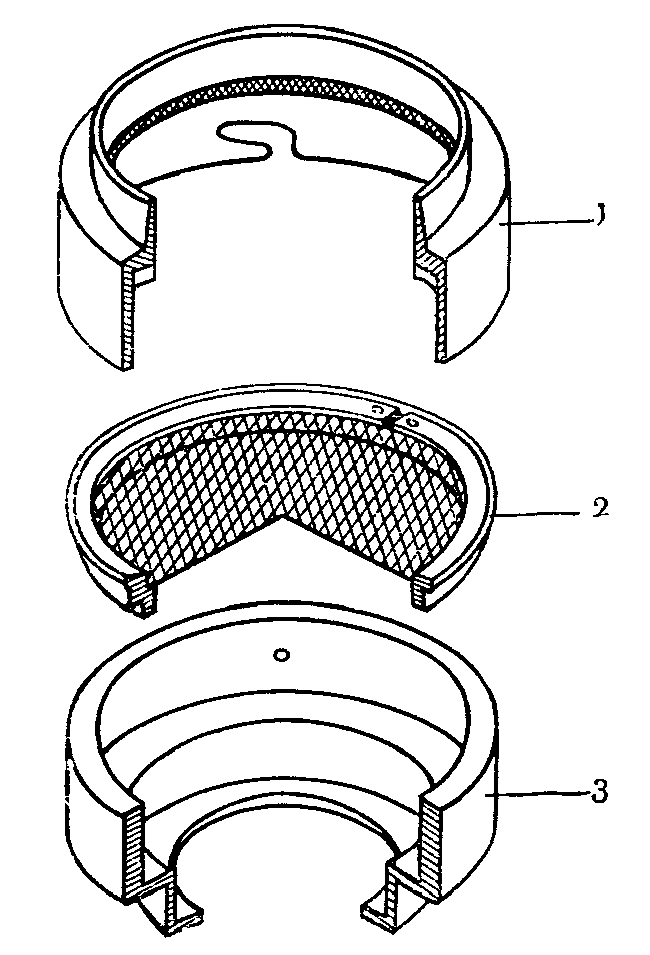
Рис 29. Патрон-насадка в разобранном виде.
1 — крышка; 2 — кассета; 3 — корпус
Разборный конусовидный стакан состоит из шести конусов, вставленных один в другой, диаметр их 11,7; 20; 25; 32; 40; 56 мм. Конусовидный стакан является средством уравнивания скорости отбора проб и скорости движения воздуха, а также для обеспечения поступления воздуха на всю рабочую поверхность фильтра, исключив возможность образования турбулентных завихрений, для чего угол корпуса не должен превышать 15°.
Для установления диаметра отверстия конуса измеряют скорость движения воздуха с помощью представленного графика (рис. 30), в котором на оси абсцисс отложена скорость ветра в метрах в секунду, а на оси ординат — соответствующие диаметры входных отверстий стаканов.
Установление диаметра стакана производится следующим образом: из точки на оси абсцисс, которая соответствует движению скорости ветра, например, 5 м/с, проводят перпендикуляр до пересечения с кривой для отбора проб со скоростью, например, 150 л/мин. Точку пересечения соединяют с осью ординат линией, параллельной оси абсцисс, и в точке соприкосновения этой линии с осью ординат будет искомый диаметр входного отверстия конусовидного стакана, в данном случае — 25 мм. Если необходимый диаметр не соответствует входному отверстию имеющихся шести диаметров стаканов, берут стакан с наиболее близким диаметром.
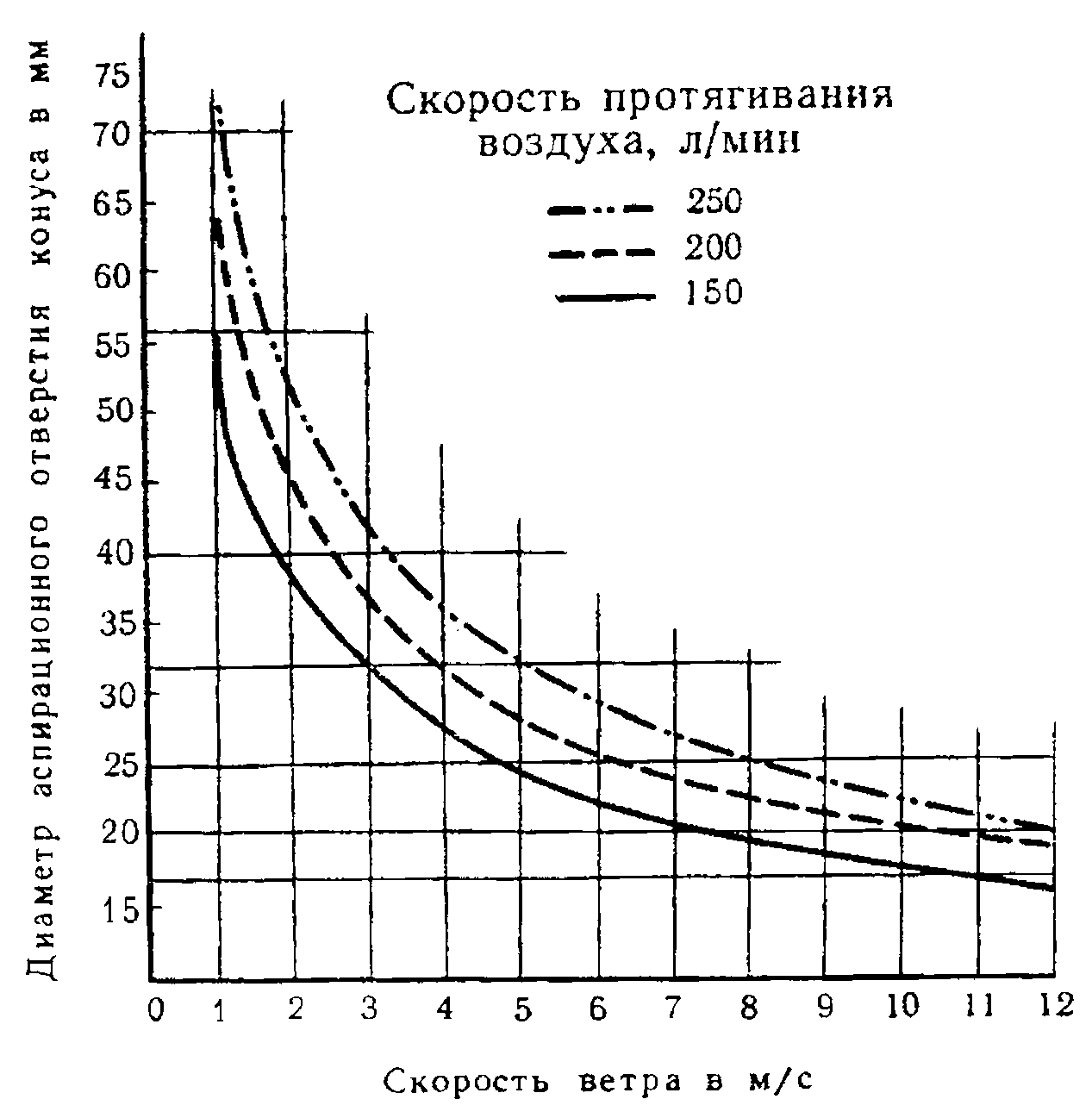
Рис. 30. График зависимости диаметра входных отверстий
конуса от скорости ветра
в) Манометр типа реометра. Шкала тарирована на производительность аспиратора в литрах в минуту. Заполняется керосином.
Отбор пробы
а) Для определения разовой концентрации воздух протягивают через фильтр из ткани ФПП-15-1,5 в течение 15 — 20 мин. со скоростью 150 — 200 л/мин., т.е. 3 м3.
б) При определении среднесуточной концентрации воздух протягивают через указанный выше фильтр 12 раз по 10 мин. со скоростью 100 л/мин. через равные интервалы.
В качестве побудителя отсоса воздуха могут быть использованы бытовые пылесосы типа «Ракета», «Уралец» и др., работающие от сети. При этом пылесос обеспечивает отбор одновременно двух параллельных проб воздуха. При работе с автомобильным аспиратором необходимо соблюдать следующую последовательность действий:
1. Разобрать патрон-насадку поворотом против часовой стрелки и снять крышку.
2. Извлечь кольцо из кассеты.
3. Вынуть фильтр из пакета.
4. Уложить фильтр на сетку кассеты с помощью пинцета и расправить края фильтра.
5. Вставить кольцо в кассету пинцетом.
6. Надеть крышку патрона-насадки и повернуть ее по часовой стрелке до упора.
7. Вставить патрон-насадку в Т-образную трубу автомобильного аспиратора.
8. Присоединить конусовидный стакан к патрону-насадке с соответствующим диаметром входного отверстия.
9. Включить аспиратор и, пользуясь дырчатой заслонкой, помещенной на противоположном конце, где находится патрон-насадка, отрегулировать протягивание воздуха по манометру.
10. Направить отверстие конуса патрона-насадки на факел выброса.
11. Отметить время начала отбора пробы.
12. После отбора пробы снять крышку патрона-насадки, пинцетом вынуть кольцо кассеты, на сетке сложить фильтр вчетверо рабочей поверхностью внутрь и уложить фильтр в пакет из кальки.
Ход анализа
В лаборатории пробы пыли помещают в весовую комнату и через 40 — 60 мин. производят взвешивание. Если отбор проб производится при большой влажности (туман или мелкий дождь), то фильтры после отбора выдерживают в эксикаторе над хлоридом кальция в течение 2 — 3 ч до постоянного веса.
Расчет анализа см. выше.
ДВУОКИСЬ КРЕМНИЯ
Двуокись кремния (кварц) представляет собой бесцветное вещество с температурой плавления 1713° и плотностью 2,62 — 2,65.
Свободная двуокись кремния может быть в кристаллической и аморфной формах. К кристаллическим минералам группы свободной двуокиси кремния относятся кварц, тридимит, кристоболит и др.; к аморфным — опал, трепел, диатомит и др. В воде и кислотах двуокись кремния не растворима, только плавиковая кислота растворяет двуокись кремния по реакции:

При сплавлении двуокиси кремния со щелочами или карбонатами образуют соли кремниевой кислоты — силикаты. При вдыхании пыли, в которой содержится кристаллическая двуокись кремния, силикаты, могут развиться такие заболевания, как силикоз, силикатоз, которые характеризуются разрастанием соединительной ткани в органах дыхания и другими изменениями.
СУММАРНОЕ ОПРЕДЕЛЕНИЕ ДВУОКИСИ КРЕМНИЯ
И СИЛИКАТОВ В ПЫЛИ [2]
Принцип определения
Метод основан на сплавлении соединений кремния с карбонатом калия — натрия и колориметрическом определении кремния в растворе по синему кремнемолибденовому комплексу.
Чувствительность определения — 1 мкг двуокиси кремния в анализируемом объеме раствора. Соединения фосфора и мышьяка не мешают определению.
Для проведения анализа требуются платиновый тигель с крышкой, тигельные щипцы с платиновыми наконечниками, агатовая ступка, платиновая или алюминиевая палочка, тигельная или муфельная печь.
Реактивы
1. Двуокись кремния SiO2 в виде чистого кварца.
2. Стандартный раствор двуокиси кремния 0,02 г тонкоизмельченной SiO2 тщательно смешивают с 0,2 г KNaCO3 и сплавляют, как указано в ходе анализа. Плав растворяют в горячей воде и раствор переносят в мерную колбу емкостью 200 мл. По охлаждении объем раствора доводят водой до метки.
Раствор, содержащий 0,1 мг/мл SiO2, сохраняют в парафинированной или полиэтиленовой склянке. Для приготовления рабочего стандартного раствора с содержанием 10 мкг/мл исходный раствор разбавляют в 10 раз водой непосредственно перед употреблением.
Стандартный раствор можно приготовить из силиката натрия Na2SiO3·9H2O (х.ч.). 0,0473 г силиката растворяют в мерной колбе в 100 мл воды. В растворе содержится 0,1 мг SiO2 в 1 мл и он сохраняется в парафинированной или полиэтиленовой склянке. Соответствующим разбавлением водой готовят раствор, содержащий 10 мкг/мл SiO2.
3. Серная кислота, 10 н. и 1 н. растворы.
4. Винная кислота, 5% раствор.
5. Аскорбиновая кислота, 1% раствор.
6. Карбонат калия — натрия KNaCO3, безводный и измельченный. Можно применить смесь безводных K2CO3 и Na2CO3, взятых в соотношении 5:4.
Отбор пробы
Исследуемый воздух со скоростью до 100 л/мин. протягивают через фильтр АФА или бумажный беззольный фильтр, помещенный в патрон <1>.
———————————
<1> Для определения можно использовать пробы пыли, причем навеска должна быть не менее 20 мг.
Ход анализа
Фильтр осторожно вынимают из патрона воронки, складывают пылью внутрь и помещают в платиновый тигель. Перед проведением анализа тигель очищают сплавлением в нем небольших количеств KNaCO3.
Тигель помещают в слегка нагретую печь, где фильтр постепенно обугливается, затем по мере повышения температуры печи его сжигают до золы. В тигель по охлаждении вносят 0,1 г карбоната натрия — калия (KNaCO3) и тщательно перемешивают с помощью алюминиевой палочки. Тигель закрывают крышкой, помещают в печь, нагретую до 800 — 900°, где смесь выдерживают до получения однородной расплавленной массы. Затем тигель вынимают из печи и по охлаждении сплав выщелачивают небольшими порциями воды. Раствор нагревают в тигле почти до кипения и сливают в мерную колбу емкостью 25 мл.
Раствор сплава осторожно подкисляют 1 н. раствором серной кислоты до слабокислой реакции на лакмус (кислотность раствора не должна быть выше 0,02 н.) и доводят водой до метки.
В колориметрическую пробирку вносят исследуемого раствора 4 мл, добавляют при встряхивании 0,1 мл раствора молибдата аммония. Через 5 мин. наливают 1 мл раствора винной кислоты и 0,1 мл раствора аскорбиновой кислоты. Спустя 20 мин. фотометрируют в кюветах с толщиной слоя 1 — 2 см при длине волны 600 нм по сравнению с контролем, который готовят одновременно и аналогично пробам. Содержание двуокиси кремния в анализируемом объеме определяют по предварительно построенному калибровочному графику. Для построения калибровочного графика готовят шкалу стандартов (табл. 107).
Таблица 107
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
|
Рабочий стандартный раствор, мл |
0 |
0,1 |
0,2 |
0,4 |
0,6 |
0,8 |
1,0 |
1,2 |
1,4 |
1,6 |
1,8 |
2,0 |
|
Вода, мл |
4 |
3,9 |
3,8 |
3,6 |
3,4 |
3,2 |
3,0 |
2,8 |
2,6 |
2,4 |
2,2 |
2,0 |
|
Содержание двуокиси кремния, мкг |
0 |
1 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
Все пробирки шкалы обрабатывают аналогично пробам, фотометрируют и строят калибровочный график. Шкалой стандартов можно пользоваться для визуального определения, ее готовят в колориметрических пробирках одновременно с пробами.
Расчет см. выше.
ОПРЕДЕЛЕНИЕ СВОБОДНОЙ ДВУОКИСИ КРЕМНИЯ В ПЫЛИ [4]
Принцип определения
Метод основан на сплавлении свободной двуокиси кремния и силикатов с бикарбонатом калия или натрия в присутствии хлоридов. При этом свободная двуокись кремния переходит в растворимую щелочную соль кремниевой кислоты, а кремниевая кислота силиката сохраняется в составе его молекулы, обогащенной щелочью плавня, и в раствор не переходит. Содержание двуокиси кремния определяют колориметрическим методом по реакции с молибденовой синью.
Чувствительность определения — 5 мкг двуокиси кремния в анализируемом объеме раствора.
Силикаты не мешают определению.
Аппаратура см. выше.
Реактивы
1. Стандартный раствор двуокиси кремния. Навеску кварца х.ч. в количестве 0,05 г тщательно растирают (до состояния пудры) в агатовой ступке, перемешивают с 2,5 г составного плавня из солей калия или с 2 г плавня из солей натрия и сплавляют в серебряном или платиновом тигле. Сплавление проводят в муфельной печи при температуре 800 — 850 °C. Однородная масса в тигле расплавляется до жидкого состояния и выдерживается при этой температуре в течение 2 — 3 мин. Затем после охлаждения остывшую массу в том же тигле обрабатывают постепенно 40 мл 5% раствора карбоната натрия при кипении. Каждую порцию раствора из тигля переводят по палочке в воронку с плотным фильтром, в мерную колбу емкостью 100 мл. Фильтрат доводят водой до 100 мл. В исходном растворе содержится 0,5 мг/мл двуокиси кремния. Рабочий раствор с содержанием 100 мкг/мл готовят путем разбавления водой исходного раствора. Стандартные растворы должны быть свежеприготовленными.
2. Бикарбонат калия или натрия, х.ч., безводный.
3. Хлорид калия или натрия х.ч., безводный.
4. Составной плавень. Равные весовые количества тонкорастертых бикарбоната и хлорида калия (или бикарбоната и хлорида натрия) тщательно перемешивают и смесь растирают в агатовой ступке небольшими порциями, которые затем соединяют вместе и снова растирают и перемешивают. Готовый составной плавень сохраняют в банке с пришлифованной пробкой.
Однородность плавня проверяют 3 раза титрованием по 1 г плавня, растворенного в 10 мл воды 1 н. раствором соляной кислоты. На 1 г плавня должно быть израсходовано 5 мл соляной кислоты. Для сплавления применяют 50-кратное количество (к навеске пробы) плавня из солей калия или 40-кратное количество плавня из солей натрия.
5. Соляная кислота, 1 н. раствор.
6. Карбонат натрия кристаллический, 5 и 10% растворы, свежеприготовленные.
7. Соляная кислота, уд. вес 1,19, разбавленная 1:3.
8. Соляная кислота, уд. вес 1,19, разбавленная 1:1.
9. Азотная кислота, концентрированная, разбавленная 1:1.
10. Хлорид аммония, 2% раствор.
11. Уксусная кислота, 10% раствор.
12. Молибдат аммония [(NH4)6Mo7O24·4H2O], 10% раствор, готовят из перекристаллизованной соли. Для перекристаллизации готовят насыщенный раствор молибдата аммония в воде, фильтруют, выливают в равный объем этилового спирта, добавляют несколько капель аммиака до явного запаха, фильтруют на воронке Бюхнера и сушат на воздухе.
13. Виннокаменная кислота, 10% раствор.
14. Сульфат натрия кристаллический, насыщенный раствор на холоду, свежеприготовленный.
Отбор пробы
Для определения свободной двуокиси кремния можно использовать пробы пыли, отобранные на бумажный фильтр или фильтр АФА-ХТ, помещенный в патрон. Скорость отбора до 100 л/мин. (для определения двуокиси кремния навеска должна быть не менее 20 мг).
Ход анализа
Фильтр с пылью взвешивают и сжигают в муфельной печи при температуре 600 °C, остаток взвешивают.
Если в пыли отсутствуют несиликатные соединения металлов (Fe, Ca, Mg и др.) — окиси, гидроокиси или карбонаты, пробу, растертую до состояния пудры в агатовой ступке, сплавляют непосредственно с плавнем. Если указанные вещества присутствуют, пробу подвергают предварительной обработке кислотами. Для этого пробу помещают в стакан и обрабатывают 10 мл смеси из равных объемов соляной и азотной кислот, разведенных 1:1, при кипении в течение 2 мин. Охлажденный раствор фильтруют и фильтр с осадком обрабатывают дважды по 10 мл нагретым до кипения 2% раствором хлорида аммония, затем 20 мл кипящего 10% раствора карбоната натрия и снова дважды по 10 мл кипящего 2% раствора хлорида аммония.
Затем фильтр с осадком подсушивают и помещают в серебряный или платиновый тигель, сжигают и прокаливают в муфельной печи. Сплавление пробы с плавнем производят следующим образом: берут навеску составного плавня 2,5 г из солей калия и 2 г из солей натрия. Из этой навески вносят небольшое количество плавня в охлажденный тигель с пробой, перемешивают палочкой и переносят в агатовую ступку. Тигель дважды обрабатывают оставшимся плавнем, который переносят в агатовую ступку и все растирают до состояния пудры. Полученную смесь снова вносят в тигель, остатком плавня обрабатывают агатовую ступку и также переносят в тигель. Содержимое тигля тщательно перемешивают и тигель помещают в муфельную печь для сплавления, постепенно нагревая печь до 800 — 900 °C. После полного расплавления массы тигель выдерживают при этой температуре еще в течение 2 — 5 мин. Для извлечения кремниевой кислоты застывший плав обрабатывают 40 мл 5% раствора карбоната натрия при нагревании; для этого небольшими порциями вносят раствор карбоната натрия в тигель, нагревают почти до кипения и фильтруют через плотный фильтр. Тигель промывают водой, которую фильтруют через тот же фильтр, и доводят объем фильтра до 100 мл дистиллированной водой. Если раствор мутный, то его фильтруют еще раз через тот же фильтр.
Для анализа в колориметрические пробирки вносят 1 и 5 мл пробы. Готовят шкалу стандартов (табл. 108).
Таблица 108
ШКАЛА СТАНДАРТОВ
|
Номер стандарта |
Контроль |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
|
Рабочий стандартный раствор, мл |
0 |
0,05 |
0,1 |
0,2 |
0,3 |
0,4 |
0,6 |
0,8 |
1 |
|
5% раствор карбоната натрия, мл |
5 |
4,95 |
4,9 |
4,8 |
4,7 |
4,6 |
4,4 |
4,2 |
4 |
|
Содержание двуокиси кремния, мкг |
0 |
5 |
10 |
20 |
30 |
40 |
60 |
80 |
100 |
Во все пробирки проб и шкалы добавляют по 0,5 мл соляной кислоты (1:3) и 0,5 мл 10% раствора уксусной кислоты, взбалтывают, приливают по 1 мл 10% раствора молибдата аммония, снова взбалтывают и оставляют на 15 мин. По истечении этого времени добавляют по 0,5 мл 10% раствора виннокаменной кислоты, взбалтывают и оставляют на 5 мин., затем приливают по 1 мл насыщенного раствора сульфита натрия, взбалтывают и ставят на 5 мин. на водяную баню, нагретую до 70 °C. Через 5 мин. пробирки охлаждают и сравнивают интенсивность окраски проб со шкалой стандартов.
Расчет
Количество двуокиси кремния (X) в процентах вычисляют по формуле:
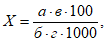
где а — количество двуокиси кремния, найденное в анализируемом объеме пробы (мкг); б — объем раствора, взятый для анализа (мл); в — общий объем пробы (мл); г — навеска пыли (мг); 1/1000 — коэффициент пересчета микрограммов в миллиграммы; 100 — коэффициент пересчета найденного количества двуокиси кремния в проценты.
ЛИТЕРАТУРА
1. Гельденскиольд Р.С., Минаев А.А. Гравиметрический метод определения пыли в атмосферном воздухе с использованием фильтра из ткани ФПП-15-1,5. Гиг. и сан., 1968, 1.
2. Определение двуокиси кремния и силикатов. В кн.: Е.А. Перегуд, Е.В. Гернет. Химический анализ воздуха промышленных предприятий. Л., «Химия», 1973.
3. Панин К.П. Определение сажи в атмосферном воздухе. Гиг. и сан., 1968, 12.
4. Полежаев Н.Г. Определение свободной двуокиси кремния в присутствии силикатов в промышленных выбросах и атмосферной пыли. Гиг. и сан., 1957, 11.
5. Рязанов В.А. Определение сажи в воздухе. В сб.: Труды Пермского медицинского института, 1934, т. VII.
УТВЕРЖДАЮ
Заместитель Главного
государственного
санитарного врача СССР
А.И.ЗАИЧЕНКО
22 декабря 1988 г. N 4945-88
МЕТОДИЧЕСКИЕ УКАЗАНИЯ ПО ОПРЕДЕЛЕНИЮ ВРЕДНЫХ ВЕЩЕСТВ В СВАРОЧНОМ АЭРОЗОЛЕ (ТВЕРДАЯ ФАЗА И ГАЗЫ)
АННОТАЦИЯ
«Методические указания по контролю воздуха при сварочных и плазменных процессах» разработаны взамен утвержденных в 1981 г. Минздравом СССР «Методических указаний на определение вредных веществ в сварочном аэрозоле (твердая фаза и газы)» N 2348-81 и в развитие ГОСТ 12.1.005-76 «ССБТ. Воздух рабочей зоны. Общие санитарно-гигиенические требования» и ГОСТ 12.1.016-79 «ССБТ. Воздух рабочей зоны. Требования к методикам измерения концентраций вредных веществ» и Методических указаний «Контроль содержания вредных веществ в воздухе рабочей зоны» N 3936-85 Минздрава СССР.
С введением настоящих Методических указаний утрачивают силу «Методические указания на определение вредных веществ в сварочном аэрозоле (твердая фаза и газы)» N 2348-81 Минздрава СССР.
Необходимость издания настоящего документа вызвана тем, что МУ N 2348-81 содержат в основном фотометрические методы, разработанные в 60-х годах, многие из которых не удовлетворяют современным требованиям, изложенным в МУ «Контроль содержания вредных веществ в воздухе рабочей зоны» N 3936-85. Кроме того, большое количество ошибок и неточностей, содержащихся в документе N 2348-81, затрудняет использование его на практике.
Внедрение новых технологий сварочных и плазменных процессов, усложнение композиций свариваемых материалов выдвигают задачу совершенствования методов санитарно-химического контроля воздуха рабочей зоны с применением современной аппаратуры.
В настоящее время получили развитие методы переменно-токовой полярографии, атомно-абсорбционной спектрофотометрии, потенциометрии с ионоселективными электродами, позволяющие значительно повысить чувствительность, селективность, точность определения и увеличить оперативность получения результатов.
Анализ оснащенности санитарно-химических лабораторий СЭС, промышленных предприятий показал, что они располагают полярографами, атомно-абсорбционными спектрофотометрами, ионоселективными электродами и др. Однако отсутствие систематизированного сборника МУ, включающего утвержденные физико-химические методы, сдерживает эксплуатацию этих приборов.
Предлагаемый документ позволяет восполнить этот пробел. В документ включено 12 новых методик взамен устаревших, остальные методики апробированы, откорректированы в соответствии с ГОСТ 12.1.016-79 и МУ N 3936-85.
Настоящие Методические указания предназначены для санитарных лабораторий промышленных предприятий и учреждений санитарно-эпидемиологической службы, осуществляющих контроль за содержанием вредных веществ в воздухе рабочей зоны, а также организаций и специалистов, проводящих работы по гигиенической оценке сварочных материалов и способов сварки, наплавки и термической резки металлов, являющихся источником выделения сварочных аэрозолей (СА), с целью проведения оздоровительных мероприятий и оценки их эффективности.
Методические указания подготовлены Киевским институтом гигиены труда и профзаболеваний (Горбань Л.Н.); Ленинградским научно-исследовательским институтом охраны труда (Буренко Т.С.); Ленинградским научно-исследовательским институтом гигиены труда и профзаболеваний (Якимова В.И.); Ордена Трудового Красного Знамени научно-исследовательским институтом гигиены труда и профзаболеваний Российской АМН (Муравьева С.И., Бабина М.Д.); Центральным научно-исследовательским институтом охраны труда (Прохорова Е.К., Зайцева З.В.).
1. ОБЩАЯ ХАРАКТЕРИСТИКА СВАРОЧНЫХ АЭРОЗОЛЕЙ
1.1. СА представляют собой сложные газо-аэрозольные смеси химических веществ, выделяющихся при дуговых, плазменных и других высокотемпературных газопламенных способах сварки, наплавки, резки и напыления металлов.
Дисперсная фаза или же твердая составляющая СА (ТССА) состоит из мельчайших частиц перенасыщенных паров металлов и других веществ, входящих в состав сварочных, присадочных, напыляемых материалов и основного металла, которые конденсируются за пределами зоны высокотемпературного нагрева.
Газовая составляющая СА (ГССА) представляет собой смесь газов, образующихся при термической диссоциации газо-шлакообразующих компонентов этих материалов (CO, CO2, HF и др.) или же за счет фотохимического действия ультрафиолетового излучения дугового разряда (плазмы) на молекулы газов воздуха (NO, NO2, O3).
1.2. Химический состав СА зависит от состава сварочных, присадочных, напыляемых материалов (электроды, проволоки, ленты, флюсы, порошки и др.), состава основного (свариваемого, направляемого либо разрезаемого) металла, режимов сварки, наплавки, резки, напыления, состава защитных газов и газовых смесей. По данным современных физико-химических исследований (рентгеноструктурного, спектрального и др. методов анализа) ТССА представляет собой сложную смесь металлов, простых и сложных оксидов металлов и шпинелей MnFe2O4, CaFe2O4, (Fe, Mn)O.Fe2O3, K2Cr2O7, Na2Cr2O7, Fe3O4 и др.), фторидов (NaF, KF, K3FeF6, K2SiF6, CaF2 и др.), силикатов (CaSiO3, -Si-O-Si-O-Si-, Fe2[SiO4], Mn2[SiO4] и др.).
1.3. Частицы ТССА — полидисперсны, имеют размеры от тысячных долей мкм до 0,4 — 0,6 мкм и более, неоднородное морфологическое строение (многослойны, многоядерны). Газы ГССА способны адсорбироваться на поверхности твердых частиц, захватываться внутрь их скоплений. При этом локальные концентрации газов, адсорбированных на частицах ТССА, могут существенно превышать их концентрации непосредственно в ГССА.
1.4. Независимо от способа высокотемпературной обработки металлов, СА могут иметь близкий химический состав и соотношение отдельных веществ — ингредиентов ТССА и ГССА. В связи с этим их целесообразно группировать в укрупненные классы газо-аэрозольных смесей относительно постоянного состава, контроль за содержанием которых в воздухе рабочей зоны допускается проводить по наиболее опасным и характерным компонентам ТССА и ГССА.
В тех случаях, когда состав известен не полностью, необходима предварительная его расшифровка для определения ведущих ингредиентов, по которым целесообразно и оправдано осуществление контроля за состоянием воздушной среды. В тех случаях, когда величина ПДК вредного вещества зависит от его процентного содержания в СА (Приложение 2, п. 12, 15), необходимо предварительно определить навеску СА на фильтре, которая должна быть не менее 5 мг.
2. ОСНОВНЫЕ ТРЕБОВАНИЯ К ОТБОРУ ПРОБ ВОЗДУХА
2.1. Отбор проб воздуха для определения уровня загрязнения воздушной среды при сварочных, наплавочных работах, резке и напылении металлов следует проводить в зоне дыхания работающих под наголовным или ручным щитом.
При измерении концентраций вредных веществ в зоне дыхания рабочих, занятых автоматическими способами сварки, наплавки и резки (контактной, под флюсом, электрошлаковой и др.) и не пользующихся защитными щитками, зоной дыхания следует считать пространство, ограниченное радиусом 50 — 60 см вокруг головы работающего.
2.2. Для характеристики общего фона загрязнения воздуха производственного помещения, где проводятся сварочные, наплавочные работы, резка и напыление металлов, отбор проб воздуха следует осуществлять в рабочей зоне на расстоянии не менее 2 м от рабочего места.
2.3. Отбор проб должен производиться при характерных производственных условиях. Любые нарушения технологического процесса (превышение либо занижение силы сварочного тока, напряжения, применение «нетипичных» сварочных и наплавочных материалов и др.) или неправильная эксплуатация оборудования и всех предусмотренных средств предотвращения загрязнения воздуха вредными веществами (устройств местной вентиляции, общеобменной вентиляции, укрытий и др.) подлежат устранению до начала проведения измерений.
2.4. Разовое определение концентраций вредных веществ должно производиться при непрерывном или последовательном отборе проб ТССА и ГССА в течение 15-минутного стандартного отрезка времени. Если чувствительность методов анализа позволяет в течение 15 минут отобрать не одну, а несколько последовательных проб, то для сопоставления с величинами ПДКм.р. концентрацию того или иного наиболее опасного и характерного вредного вещества, выделяющегося в составе ТССА и/или ГССА, следует находить как среднюю величину из результатов измерений, выполненных за указанный период времени.
Для вредных веществ, метод определения которых не позволяет обнаружить 0,5 ПДКм.р. за 15 минут отбора пробы, допускается увеличение времени отбора, но не более чем до 30 мин.
Допустимая объемная скорость отбора проб воздуха на фильтры АФА из подручного или наголовного щитка составляет 10 л/мин.
2.5. Отбор проб ТССА осуществляется на аналитические аэрозольные фильтры АФА-ХП, АФА-ВП или АФА-ХА с объемным расходом 10 — 15 л/мин. Тип фильтра, применяемого для концентрирования компонентов ТССА, определяется ходом последующего химического анализа и должен строго соблюдаться. В случаях, когда материал фильтра на ход анализа не влияет, в соответствующих разделах методик тип фильтра не указывается.
Отбор проб ГССА проводится с концентрированием в жидкостные поглотительные приборы, сорбционные трубки либо без концентрирования в медицинские шприцы или пипетки.
2.6. Если стадия технологического процесса (операции) непродолжительна и не позволяет отобрать пробу воздуха за один цикл (расплавление одного электрода, «прихватка» деталей и т.д.), отбор пробы воздуха на этот же фильтр или в один и тот же поглотитель необходимо продолжить при повторении операции.
2.7. Для получения достоверных результатов при санитарно-гигиенических исследованиях воздушной среды на каждом обследуемом рабочем месте сварщика, наплавщика, резчика металлов, операторов установок напыления порошков металлов должно быть последовательно отобрано не менее 5 проб воздуха для определения концентраций ведущего токсического ингредиента ТССА и не менее 5 проб наиболее характерного токсического ингредиента ГССА.
Средние величины из результатов выполненных измерений и их доверительный интервал следует находить с учетом требований Методических указаний «Контроль содержания вредных веществ в воздухе рабочей зоны» N 3936-85 Минздрава СССР.
2.8. Периодичность санитарного контроля за соблюдением гигиенических требований к качеству воздушной среды при выполнении сварочных, наплавочных и газорезательных работ определяется по согласованию с территориальными учреждениями санитарно-эпидемиологической службы с учетом Методических указаний «Контроль содержания вредных веществ в воздухе рабочей зоны» N 3936-85 Минздрава СССР и результатов предшествующих измерений.
2.9. Санитарный контроль воздуха рабочей зоны при сварочных, наплавочных работах, а также резке и напылении металлов, сопровождающихся выделением вредных веществ, относящихся к I и II классам опасности, следует осуществлять с помощью физико-химических методов анализа. Гравиметрический метод контроля воздуха рабочей зоны допускается в случаях загрязнения его ТССА, состоящей из веществ, относящихся к III и IV классам опасности (TiO2, окислы железа и др.), а также при оперативном контроле эффективности работы средств вентиляции по согласованию с учреждениями санитарно-эпидемиологической службы.
2.10. Для наиболее опасных и характерных вредных веществ — ингредиентов ТССА и ГССА, которые имеют соответствующую среднесменную ПДК (ПДКс.с.), допускается осуществлять контроль путем измерения среднесменных концентраций.
Для характеристики уровня среднесменных концентраций, воздействующих на рабочих-сварщиков, наплавщиков, резчиков металлов, а также обслуживающих установки для напыления металлов, занятых однотипными производственными операциями (с использованием одних и тех же электродов, проволок одного и того же диаметра, флюсов и др.; при сварке, наплавке и резке одних и тех же металлов и пр.), необходимо проводить обследование не менее 5 человеко-смен. Расчет среднесменных концентраций производится в соответствии с Методическими указаниями «Контроль содержания вредных веществ в воздухе рабочей зоны» N 3936-85 Минздрава СССР.
3. МЕТОДЫ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ
3.1. ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Раздельное измерение концентрации железа, никеля, марганца, титана и оксидов хрома (III и VI)
Характеристика метода
Определение основано на колориметрических реакциях отдельных металлов с органическими реагентами.
Отбор проб воздуха проводится с концентрированием на фильтр.
Основные метрологические характеристики методик измерения концентраций приведены при описании определения каждого металла.
Определение отдельных металлов проводят в аликвотных частях раствора плава.
Время подготовки проб к определению 5 — 6 часов, включая отбор проб 20 минут. Время самого определения указано в каждой методике отдельно.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56 М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Тигли фарфоровые, ГОСТ 9147-80Е.
Тигли платиновые, ГОСТ 6563-75.
Щипцы тигельные.
Ступка фарфоровая, ГОСТ 9147-80Е.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 500, 1000 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 и 50 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 0,2, 1, 2,5 и 10 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Стаканы химические, ГОСТ 25336-82Е.
Воронки стеклянные, ГОСТ 25336-82Е.
Реактивы, растворы, материалы
Натрий углекислый (карбонат натрия), ГОСТ 83-79, хч.
Калий азотнокислый (нитрат калия), ГОСТ 4217-77, хч.
Кислота серная, ГОСТ 4204-77, хч, 10% раствор (по объему).
Плавень: смешивают две части карбоната натрия и одну часть нитрата калия. Смесь растирают в фарфоровой ступке. Плавень хранят в банке с притертой пробкой.
Фильтры АФА-ХП, АФА-ВП или АФА-ХА, ТУ 95.743-80.
Фильтры обеззоленные «синяя лента», ГОСТ 12026-76.
Отбор пробы воздуха
Воздух с объемным расходом 5 — 15 л/мин. аспирируют через фильтр АФА. Пробы не следует хранить из-за возможных потерь шестивалентного хрома. Для определения перечисленных металлов на уровне 1/2 ПДК следует отобрать 200 л воздуха.
Подготовка пробы к раздельному измерению компонентов
Определение растворимого оксида хрома (VI) производят в водном фильтрате. Для этого фильтр с отобранной пробой помещают на чистый обеззоленный фильтр «синяя лента», вложенный в воронку, смачивают этиловым спиртом (0,2 — 0,3 мл) и обрабатывают 10 мл теплой (40 — 50 град. C) воды.
Для определения 3-валентного хрома, марганца, железа, титана и никеля фильтр АФА после обработки его водой и фильтр «синяя лента», через который фильтровали исследуемую пробу, помещают в фарфоровый тигель, подсушивают на воздухе и ставят в холодную муфельную печь, постепенно повышая температуру до 750 — 800 град. C.
После озоления фильтров тигель вынимают из муфельной печи и остаток по охлаждении тщательно смешивают и растирают лопаточкой с 0,5 — 1,0 г плавня. Далее тигель помещают в охлажденный до 300 — 400 град. C муфель, снова повышают температуру по 800 — 850 град. C и оставляют в нем тигель на 25 — 30 минут до полного сплавления смеси. По охлаждении тигля плав обрабатывают 25 мл раствора серной кислоты под тягой до полного растворения.
Примечание. При использовании фарфоровых тиглей разных партий возможно появление мути в контрольных растворах плавня за счет различного состава фарфора и качества глазури. В связи с этим обязательна постановка контрольных опытов для каждой партии тиглей с использованием чистых фильтров (не менее 5 — 7 тиглей). В случае, если оптическая плотность распоров превышает 0,04 — 0,05, партия тиглей бракуется. Для анализа могут быть использованы также платиновые или кварцевые тигли.
Измерение концентрации оксида хрома (VI)
Характеристика метода
Определение основано на реакции взаимодействия шестивалентного хрома с дифенилкарбазидом в кислой среде с образованием соединения, окрашенного в красно-фиолетовый цвет.
Нижний предел измерения содержания оксида хрома (VI) в фотометрируемом растворе составляет 0,5 мкг.
Нижний предел измерения в воздухе 0,003 мг/куб. м (при отборе 200 л воздуха).
Диапазон измеряемых концентраций от 0,003 до 0,06 мг/куб. м.
Измерению не мешает марганец. Мешают железо при содержании более 1 мг и молибден в количестве более 8 мг.
Суммарная погрешность измерения не превышает +/- 10%.
Время выполнения измерения 20 — 25 мин.
Реактивы, растворы и материалы
Калий двухромовокислый, ГОСТ 4220-75, хч.
Кислота уксусная ледяная, ГОСТ 61-75, хч.
Этиловый спирт, ГОСТ 18300-72, хч или ГОСТ 5963-67.
Дифенилкарбазид, ГОСТ 5859-78, чда, 0,5% раствор, 0,1 г дифенилкарбазида растворяют в 2,0 мл ледяной уксусной кислоты и прибавляют 20 мл этилового спирта.
Стандартный раствор оксида хрома (VI) N 1 с концентрацией 1 мг/мл готовят растворением в воде 0,1471 г двухромовокислого калия в мерной колбе вместимостью 1 л. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор оксида хрома (VI) N 2 с концентрацией 10 мкг/мл готовят путем разбавления 2 мл стандартного раствора N 1 водой в мерной колбе вместимостью 200 мл. Применяют свежеприготовленный раствор.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х час.) готовят согласно таблице 1.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ХРОМОВОГО АНГИДРИДА
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание оксида хрома (VI) в градуировочном растворе, мкг |
| 1 | 0 | 10 | 0 |
| 2 | 0,05 | 9,95 | 0,5 |
| 3 | 0,1 | 9,90 | 1,0 |
| 4 | 0,2 | 9,80 | 2,0 |
| 5 | 0,4 | 9,60 | 4,0 |
| 6 | 0,6 | 9,40 | 6,0 |
| 7 | 0,8 | 9,20 | 8,0 |
| 8 | 1,0 | 9,00 | 10,0 |
Во все пробирки шкалы добавляют по 1 мл 0,5% раствора дифенилкарбазида, взбалтывают и выдерживают 15 минут. Затем измеряют оптическую плотность при длине волны 540 нм в кюветах с толщиной поглощающего слоя 2 см по отношению к контрольному раствору (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности градуировочных растворов от содержания оксида хрома (VI) (мкг), проверку которого проводят 1 раз в квартал в случае использования новой партии реактивов.
Проведение измерения
9 мл водного фильтрата вносят в колориметрические пробирки и доводят объем пробы дистиллированной водой до 10 мл. Далее измерение идет аналогично приготовлению градуировочных растворов. Оптическую плотность измеряют по сравнению с контролем, который готовят одновременно и аналогично пробам.
Содержание оксида хрома (VI) в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию оксида хрома (VI) в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации оксида хрома (III)
Характеристика метода
Определение основано на окислении трехвалентного хрома персульфатом аммония до шестивалентного хрома. После разрушения избытка персульфата аммония определение Cr (VI) проводят по реакции с дифенилкарбазидом.
Нижний предел измерения оксида хрома (VI) в фотометрируемом объеме раствора 0,5 мкг, оксида хрома (III) 0,4 мкг.
Нижний предел измерения оксида хрома (III) в воздухе — 0,5 мг/ куб. м (при отборе 200 л).
Диапазон измеряемых концентраций от 0,5 до 9,5 мг/куб. м.
Измерению не мешают сопутствующие металлы.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 3 часа.
Реактивы, растворы и материалы
Аммоний надсернокислый (персульфат аммония), ГОСТ 20478-75, хч.
Серебро азотнокислое, ГОСТ 1277-75 хч, 2% раствор.
Остальные реактивы и растворы приведены выше.
Проведение измерения
В коническую колбу вносят 10 мл раствора пробы в 10% серной кислоте, добавляют 0,5 мл раствора нитрата серебра и 0,2 г персульфата аммония. Колбу ставят на газовую горелку с сеткой или электроплитку и интенсивно кипятят 20 — 25 минут. По мере испарения в колбу доливают дистиллированную воду так, чтобы объем жидкости был 10 — 15 мл. Цвет жидкости становится сначала желтым (окисление хрома), а затем розовым (окисление марганца). После полного разрушения персульфата аммония в колбу добавляют по милям раствор соляной кислоты для разрушения марганцевой кислоты. Хромовая кислота при этом не разрушается. Раствор соляной кислоты следует добавлять несколько раз по 0,3 — 0,5 мл до исчезновения розового оттенка. После разрушения марганцевой кислоты раствор охлаждают и количественно переносят в мерную колбу вместимостью 100 мл, многократно промывая коническую колбу дистиллированной водой и собирая промывные воды в ту же мерную колбу, объем которой доводят до метки водой.
Полученный раствор в количестве 1,0 мл вносят в колориметрическую пробирку, доводят до 10 мл водой и далее обрабатывают и фотометрируют аналогично градуировочным растворам для измерения концентрации оксида хрома (VI).
Содержание оксида хрома (VI) в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию оксида хрома (III) в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета оксида хрома (VI) на оксид хрома (III) — 0,76.
Измерение концентрации марганца
Характеристика метода
Определение основано на реакции окисления соединений марганца персульфатом аммония в присутствии нитрата серебра как катализатора с образованием марганцевой кислоты, окрашенной в малиново-красный цвет.
Нижний предел измерения содержания марганца в объеме анализируемого раствора 1,0 мкг.
Нижний предел измерения в воздухе 0,05 мг/куб. м (при отборе 200 л воздуха) <*>.
<*> Расчет предела обнаружения проведен с учетом значения ПДК для Mn — 0,1 мг/куб. м.
Диапазон измеряемых концентраций для марганца от 0,05 мг/куб. м до 1,25 мг/куб. м.
Определению марганца мешает железо, влияние которого устраняют добавлением ортофосфорной кислоты. Определению не мешает хром.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 1,5 часа.
Реактивы, растворы и материалы
Марганец сернокислый, 5-водный, ГОСТ 435-77, чда.
Серебро азотнокислое, ГОСТ 1277-75, хч, 2% раствор.
Аммоний надсернокислый, ГОСТ 20478-75, хч, 10% раствор, свежеприготовленный.
Кислота ортофосфорная, ГОСТ 6552-58, хч, 50% раствор.
Кислота серная, ГОСТ 4204-77, хч, 10% раствор.
Стандартный раствор N 1, содержащий 100 мкг/мл марганца, растворяют 0,0439 г 5-водного сернокислого марганца в мерной колбе вместимостью 200 мл в 10% растворе серной кислоты. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор N 2 с концентрацией марганца 10 мкг/мл готовят разведением раствора N 1 10% раствором серной кислоты в 10 раз. Раствор свежеприготовленный.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблице 2.
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МАРГАНЦА
| N стандарта | Стандартный раствор N 2, мл | Раствор серной кислоты, 10%, мл | Содержание марганца в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 1,0 |
| 3 | 0,2 | 4,8 | 2,0 |
| 4 | 0,4 | 4,6 | 4,0 |
| 5 | 0,6 | 4,4 | 6,0 |
| 6 | 0,8 | 4,2 | 8,0 |
| 7 | 1,0 | 4,0 | 10,0 |
Во все пробирки шкалы прибавляют по 0,2 мл 2% раствора нитрата серебра и по 1 мл растворов 10% персульфата аммония и 50% ортофосфорной кислоты. После прибавления каждого реактива растворы взбалтывают и погружают в кипящую баню на 5 — 10 минут.
По охлаждении растворов измеряют величину оптической плотности при 545 нм в кюветах с толщиной слоя 1 см по сравнению с контрольным раствором (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности растворов от содержания марганца в градуировочном растворе (мкг), проверку которого проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
1 — 2,5 мл раствора пробы в 10% серной кислоте вносят в колориметрические пробирки, доводят до 5 мл кислотой и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание марганца в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию марганца в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации железа
Характеристика метода
Определение основано на реакции взаимодействия ионов железа с сульфосалициловой кислотой в аммиачной среде с образованием окрашенного соединения.
Нижний предел измерения содержания железа в объеме анализируемого раствора 1 мкг.
Нижний предел измерения железа в воздухе 1,5 мг/куб. м (при отборе 200 л воздуха).
Диапазон измеряемых концентраций от 1,5 до 15 мг/куб. м.
Определению не мешают молибден, ванадий, хром, марганец. Мешают кобальт, никель в количествах более 1,2 мг, медь в количествах более 0,2 мг.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 40 минут.
Реактивы, растворы и материалы
Железо металлическое, ТУ 6-09-3000-78, осч или ТУ 6-09-2227-81, ч.
Серная кислота, ГОСТ 4204-77, хч, 10% раствор.
Сульфосалициловая кислота, двуводная, ГОСТ 4478-78, чда, 10% раствор.
Соляная кислота, ГОСТ 3118-77, хч, раствор 1:1.
Азотная кислота, ГОСТ 4461-77, хч.
Аммиак водный, ГОСТ 3760-79, хч.
Основной раствор железа с концентрацией 1 мг/мл. 1,0000 г железа растворяют в термостойком химическом стакане вместимостью 50 — 100 мл в смеси 20 мл соляной кислоты 1:1 и 5 мл азотной кислоты. Раствор осторожно нагревают на плитке, не допуская разбрызгивания. Приливают в стаканы 10 мл дистиллированной воды и кипятят раствор до удаления паров оксида азота (IV). По охлаждении раствор количественно переносят в мерную колбу вместимостью 1 л путем многократного промывания стакана дистиллированной водой. Объем раствора доводят до метки водой.
Стандартный раствор железа N 1 с концентрацией 100 мкг/мл готовят путем соответствующего разбавления основного раствора 10% серной кислотой в мерной колбе.
Стандартный раствор железа N 2 с концентрацией 10 мкг/мл готовят разбавлением раствора N 1 в 10 раз 10% серной кислотой. Применяют свежеприготовленным.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение суток) готовят согласно таблице 3.
Таблица 3
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЖЕЛЕЗА
| N стандарта | Стандартный раствор N 2, мл | Раствор серной кислоты, 10%, мл | Содержание железа в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 1,0 |
| 3 | 0,2 | 4,8 | 2,0 |
| 4 | 0,4 | 4,6 | 4,0 |
| 5 | 0,6 | 4,4 | 6,0 |
| 6 | 0,8 | 4,2 | 8,0 |
| 7 | 1,0 | 4,0 | 10,0 |
Во все пробирки шкалы прибавляют по 0,5 мл 10% раствора сульфосалициловой кислоты, взбалтывают, добавляют по 1 мл концентрированного раствора аммиака, снова взбалтывают и измеряют оптическую плотность растворов при длине волны 420 — 430 нм в кюветах с толщиной слоя 1 см по сравнению с контрольным раствором (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности от содержания железа в градуировочном растворе (мкг), проверку которого проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
1 мл раствора пробы в 10% серной кислоте вносят в мерную колбу вместимостью 25 мл и доводят до метки 10% серной кислотой, 2 мл полученного раствора вносят в колориметрическую пробирку, доводят объем до 5 мл кислотой и далее обрабатывают и фотометрируют аналогично градуировочным растворам. Содержание железа в анализируемом объеме раствора пробы находят по градуировочному графику.
Расчет концентрации
![]() , мг/куб. м,
, мг/куб. м,
где:
а — содержание железа в анализируемом объеме раствора пробы, найденное по градуировочному графику, мкг;
V — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л;
К — коэффициент пересчета железа на оксид железа — 1,43.
Измерение концентрации никеля
Характеристика метода
Фотометрическое определение основано на реакции взаимодействия иона никеля с диметилглиоксимом в щелочной среде в присутствии окислителя с образованием комплексного соединения, окрашенного в розово-коричневый цвет.
Нижний предел измерения содержания никеля в объеме анализируемого раствора 1,0 мкг.
Нижний предел измерения в воздухе 0,025 мг/куб. м (при отборе 200 л воздуха).
Диапазон измеряемых концентраций от 0,025 до 1,25 мг/куб. м.
Определению никеля не мешают железо, медь и кобальт в количествах, меньших 0,2 мг.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 40 — 45 минут.
Реактивы, растворы, материалы
Никель сернокислый, 7-водный, ГОСТ 4465-74, хч.
Кислота серная, ГОСТ 4204-77, хч, 10% раствор.
Аммоний надсернокислый (персульфат аммония), ГОСТ 20478-75, хч, 3% раствор, свежеприготовленный.
Натрий лимоннокислый, 5,5-водный, ГОСТ 22280-76, чда, 20% раствор.
Натрия гидроксид, ГОСТ 4328-77, хч, 5% и 40% растворы.
Диметилглиоксим, ГОСТ 5828-77, чда, 1% раствор в 5% растворе гидроксида натрия.
Спирт этиловый, ГОСТ 18300-72, хч или ГОСТ 5963-67.
Аммиак водный, ГОСТ 3760-79, хч, 5% и 25% растворы.
Хлороформ, ТУ 6-09-06-800-76, хч.
Бумага лакмусовая, красная.
Стандартный раствор никеля N 1 с концентрацией 100 мкг/мл готовят путем растворения 0,0478 г сернокислого никеля в 10% растворе серной кислоты в мерной колбе вместимостью 100 мл. Устойчив 2 месяца.
Стандартный раствор никеля N 2 с концентрацией 10 мкг/мл готовят соответствующим разбавлением раствора N 1 10-процентной серной кислотой. Применяют свежеприготовленный раствор.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблице 4.
Таблица 4
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ НИКЕЛЯ
| N стандарта | Стандартный раствор N 2, мл | Серная кислота, 10% раствор, мл | Содержание никеля в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 1,0 |
| 3 | 0,2 | 4,8 | 2,0 |
| 4 | 0,4 | 4,6 | 4,0 |
| 5 | 0,6 | 4,4 | 6,0 |
| 6 | 0,8 | 4,2 | 8,0 |
| 7 | 1,0 | 4,0 | 10,0 |
Во все пробирки шкалы добавляют по 0,4 мл раствора лимоннокислого натрия, 0,2 мл персульфата аммония, нейтрализуют 40% раствором едкого натра при перемешивании до слабощелочной реакции по лакмусовой бумаге, прибавляют 0,5 мл раствора диметилглиоксима и взбалтывают. Через 15 минут измеряют оптическую плотность градуировочных растворов при длине волны 530 нм в кюветах с толщиной слоя 1 см относительно контрольного раствора (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности от содержания никеля в градуировочном растворе (мкг), проверку графика проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
1 — 5 мл раствора пробы в 10% серной кислоте переносят в колориметрические пробирки, доводят объем до 5 мл раствором серной кислоты и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
При наличии в пробе железа, меди и кобальта в количествах, превышающих 0,2 мг, никель необходимо отделять. Для этого в делительную воронку отбирают 5 — 10 мл раствора пробы, нейтрализуют его 25% раствором аммиака до слабощелочной реакции по индикаторной бумаге, приливают 2 мл 1% раствора диметилглиоксима, 5 мл хлороформа и встряхивают 30 секунд. Хлороформенный слой, содержащий диметилглиоксиматы никеля и кобальта, отделяют. В воронку вводят еще 5 мл хлороформа и повторяют экстракцию. Хлороформенные экстракты объединяют и промывают в делительной воронке дважды 20 — 30 мл 5% раствора аммиака для удаления частично извлеченной меди.
Реэкстрагируют никель путем двукратной обработки хлороформенной вытяжки 5 мл 10% азотной кислоты. При этом комплекс разлагается и никель переходит в раствор кислоты.
1 — 5 мл полученного раствора вносят в пробирку, доводят объем до 5 мл азотной кислотой. Шкалу градуировочных растворов в этом случае также готовят с использованием азотной кислоты. Далее пробу обрабатывают и фотометрируют, как описано выше, относительно контрольного раствора, который готовят одновременно и аналогично пробе.
Содержание никеля в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию никеля в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации титана
Характеристика метода
Определение основано на реакции взаимодействия титана с пероксидом водорода в кислой среде.
Нижний предел измерения содержания титана в объеме анализируемого раствора 10 мкг.
Нижний предел измерения титана в воздухе 6,0 мг/куб. м (при отборе 200 л воздуха).
Диапазон измеряемых концентраций в воздухе от 6 до 62 мг/куб. м.
Определению титана не мешают железо, марганец, хром. Определению мешает ванадий, который отделяют в процессе анализа.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 20 — 25 минут.
Реактивы, растворы, материалы
Титана диоксид, ТУ 6-09-2166-77, чда.
Натрий вольфрамовокислый (вольфрамат натрия), 2-водный, ГОСТ 18289-78.
Калий сернокислый пиро (пиросульфат калия), ГОСТ 5713-75, чда.
Кислота серная, ГОСТ 4204-77, хч, 10% раствор.
Перекись водорода, ГОСТ 10929-76, хч, 3% раствор.
Натрия гидроксид, ГОСТ 4328-77, хч, 5% и 30% растворы.
Стандартный раствор, содержащий 100 мкг/мл титана, готовят из диоксида титана, предварительно прокаленного до постоянного веса. 0,1668 г двуокиси титана сплавляют с 1 — 1,5 г пиросульфата калия. Температуру печи постепенно повышают до 500 град. C и выдерживают при этой температуре 1 ч. Плав выщелачивают раствором 10% серной кислоты, переводят в мерную колбу вместимостью 1 л и доводят до метки этим же раствором. Устойчив в течение 2 месяцев.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение часа) готовят согласно таблице 5.
Таблица 5
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ТИТАНА
| N стандарта | Стандартный раствор, мл | Раствор серной кислоты, 10%, мл | Содержание титана в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 10,0 |
| 3 | 0,2 | 4,8 | 20,0 |
| 4 | 0,4 | 4,6 | 40,0 |
| 5 | 0,6 | 4,4 | 60,0 |
| 6 | 0,8 | 4,2 | 80,0 |
| 7 | 1,0 | 4,0 | 100,0 |
Во все пробирки шкалы добавляют по 1 мл 3% раствора перекиси водорода, взбалтывают и через 50 — 10 мин. фотометрируют при длине волны 460 — 480 нм в кюветах с толщиной слоя в 1 см по отношению к контролю (раствор N 1 по таблице).
Строят градуировочный график зависимости оптической плотности растворов от содержания титана в градуировочном растворе (мкг), проверку которого проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
5 мл сернокислого раствора пробы вносят в мерную колбу вместимостью 25 мл и доводят до метки 10% серной кислотой. 1 мл полученного раствора вносят в колориметрическую пробирку, доводят объем до 5 мл 10% серной кислотой и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Если в пробе присутствует ванадий, который мешает определению титана, его необходимо отделить. Для этого к 5 мл разбавленного сернокислого раствора пробы прибавляют 30% раствор едкого натра (для осаждения железа и титана). Раствор нагревают на водяной бане и фильтруют. В фильтрате определяют ванадий по реакции с вольфраматом натрия (если это надо), а осадок на фильтре промывают 5% раствором едкого натра до отрицательной реакции на ванадий, растворяя его в растворе серной кислоты.
В растворе определяют титан по реакции с перекисью водорода, как описано выше.
Расчет концентрации
![]() , мг/куб. м,
, мг/куб. м,
где:
а — содержание титана в фотометрируемом растворе пробы, найденное по градуировочному графику, мкг;
V — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
Измерение концентрации меди
Характеристика метода
Определение основано на реакции взаимодействия меди с диэтилдитиокарбаматом натрия в щелочной среде с образованием окрашенного соединения.
Отбор проб проводится с концентрированием на фильтр АФА.
Нижний предел измерения содержания меди в объеме анализируемого раствора 1 мкг.
Нижний предел измерения вещества в воздухе 0,4 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,4 до 8,0 мг/куб. м.
Определению мешает железо, влияние которого устраняется в процессе анализа добавлением динатриевой соли этилендиаминтетрауксусной кислоты.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 2,5 часа, включая отбор проб 2 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня водяная.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 100, 500 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 и 100 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75, высотой 150 мм, внутренним диаметром 15 мм.
Реактивы, растворы, материалы
Медь сернокислая, 5-водная, ГОСТ 4165-78, хч.
Аммиак водный, ГОСТ 3760-79, чда, концентрированный 25% раствор.
Аммоний хлористый, ГОСТ 3737-72, хч.
Хлоридно-аммиачный буферный раствор. 100 г хлорида аммония растворяют в небольшом количестве воды, раствор переносят в мерную колбу вместимостью 500 мл, добавляют 100 мл концентрированного раствора аммиака и доводят объем до метки водой.
Этилендиаминтетрауксусной кислоты динатриевая соль, двуводная (Трилон Б), ГОСТ 10652-73, хч, 5% раствор.
Диэтилдитиокарбамат натрия, 3-водный, ГОСТ 8864-71, чда, 5% водный раствор. Применяют свежеприготовленным. Хранят в темной склянке.
Кислота азотная, ГОСТ 4461-77, чда, 10% раствор.
Стандартный раствор меди N 1 с концентрацией 100 мкг/мл готовят растворением 0,393 г сульфата меди (чистые, не выветренные кристаллы) в дистиллированной воде в мерной колбе вместимостью 1 л. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор меди N 2 с концентрацией 10 мкг/мл готовят соответствующим разбавлением стандартного раствора N 1 водой. Применяют свежеприготовленный раствор.
Фильтры типа АФА-ХП, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА-ХП. Для измерения 1/2 ПДКм.р. меди достаточно отобрать 20 л воздуха. Пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 1 часа) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МЕДИ
| N стандарта | Стандартный раствор N 2, мл | Вода дистиллированная, мл | Содержание меди в градуировочном растворе, мкг |
| 1 | 0 | 3,0 | 0 |
| 2 | 0,1 | 2,9 | 1,0 |
| 3 | 0,2 | 2,8 | 2,0 |
| 4 | 0,5 | 2,5 | 5,0 |
| 5 | 1,0 | 2,0 | 10,0 |
| 6 | 1,4 | 1,6 | 14,0 |
| 7 | 1,6 | 1,4 | 16,0 |
| 8 | 2,0 | 1,0 | 20,0 |
Во все пробирки приливают по 0,7 мл буферного раствора, 0,2 мл раствора Трилона Б и 0,1 мл раствора диэтилдитиокарбамата натрия. После прибавления каждого реактива растворы осторожно перемешивают. Окрашенные растворы фотометрируют через 10 минут при длине волны 436 нм в кюветах с толщиной слоя 1 см относительно контроля (раствор N 1 шкалы стандартов).
Строят градуировочный график зависимости оптической плотности от содержания меди (мкг), проверку которого проводят 1 раз в квартал или при использовании новой партии реактивов.
Проведение измерения
Фильтр АФА помещают в химический стакан и заливают 10 мл 10% раствора азотной кислоты, раствор нагревают на кипящей водяной бане в течение 7 — 10 минут. Фильтр отжимают на стенке стакана стеклянной палочкой, промывают трижды 2 — 3 мл горячей воды и отбрасывают. Раствор выпаривают на кипящей водяной бане досуха. Приливают в стакан 3 — 5 мл воды и вновь упаривают досуха. Сухой остаток растворяют в 10 — 15 мл дистиллированной воды, фильтруют, если есть необходимость, переносят раствор количественно в мерную колбу вместимостью 25 мл и доводят до метки водой.
3 мл полученного раствора вносят в колориметрическую пробирку и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание меди в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию меди в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации цинка и оксида цинка
Характеристика метода
Определение основано на реакции взаимодействия тетрароданоцинката ![]() с катионом родамина «C» с образованием окрашенного соединения.
с катионом родамина «C» с образованием окрашенного соединения.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания цинка в объеме анализируемого раствора 1 мкг.
Нижний предел измерения цинка в воздухе 0,25 мг/куб. м (при отборе 10 л воздуха).
Диапазон измеряемых концентраций от 0,25 до 10,0 мг/куб. м.
Определению не мешают свинец (1:10), никель (1:100), марганец (1:100), молибден (1:50), ванадий (1:10), алюминий (1:100). Влияние железа (III) (1:5) и меди (1:10) устраняется в процессе анализа.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения определения 40 минут, включая отбор проб 5 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 100 и 500 мл.
Пипетки, ГОСТ 29292-74Е, вместимостью 1, 2, 5 и 10 мл.
Пробирки, ГОСТ 10515-75, высотой 120 мм и диаметром 15 мм.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 мл.
Чашки фарфоровые, ГОСТ 9147-80Е.
Воронки стеклянные, ГОСТ 25336-82Е, диаметром 56 мм.
Баня водная.
Реактивы, растворы, материалы
Цинк металлический, гранулированный, ГОСТ 3640-79, в.ч.
Кислота соляная, ГОСТ 3118-77, хч, 1 М раствор.
Кислота серная, ГОСТ 4204-77, хч, 0,1 н. раствор.
Гидроксиламин солянокислый, ГОСТ 5456-65, чда, 20% раствор, свежеприготовленный (хранят в склянке из темного стекла).
Тиомочевина, ГОСТ 6344-73, хч, 10% раствор, 10 г реактива растворяют в 50 мл горячей дистиллированной воды, после охлаждения объем доводят водой до 100 мл.
Калий роданистый, ГОСТ 41399-75, хч, 20% раствор.
Родамин «C» (диэтил-мета-аминофенолфталеин), ТУ РУ 856-53, 0,2% раствор.
Натрий уксуснокислый, 3-водный, ГОСТ 199-78, 15% раствор.
Кислота уксусная, ГОСТ 61-75, хч, ледяная.
Стандартный раствор N 1 с содержанием 100 мкг/мл цинка готовят растворением навески 0,5000 г цинка металлического в 5 мл 1 М соляной кислоты при нагревании, раствор охлаждают до 20 град. C, переносят в мерную колбу вместимостью 500 мл и разбавляют до метки водой. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор N 2 с содержанием 10 мкг/мл цинка готовят соответствующим разбавлением основного раствора дистиллированной водой, свежеприготовленный.
Фильтры АФА-ХП-20 или АФА-ВП-20, ТУ 95-743-80.
Бумага индикаторная универсальная, ТУ 6-09-1181-76.
Отбор проб воздуха
Воздух с объемным расходом 5 л/мин. аспирируют через фильтр АФА.
Для определения 1/2 ПДК окиси цинка достаточно отобрать 10 л воздуха. Пробы хранятся в течение 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЦИНКА
| N стандарта | Стандартный раствор N 2, мл | Вода дистиллированная, мл | Содержание цинка в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 1,0 |
| 3 | 0,2 | 4,8 | 2,0 |
| 4 | 0,3 | 4,7 | 3,0 |
| 5 | 0,5 | 4,5 | 5,0 |
| 6 | 0,7 | 4,3 | 7,0 |
| 7 | 1,0 | 4,0 | 10,0 |
Во всех пробирках шкалы устанавливают pH раствора 3 — 4, добавляя по каплям 0,1 н. серную кислоту (по индикаторной бумаге). Приливают 1 мл раствора гидроксиламина, затем через 2 минуты — 1 мл раствора ацетатного буфера, 1 мл раствора тиомочевины и 1 мл раствора роданида калия. После добавления каждого реактива раствор тщательно перемешивают. Доводят объем до 10 мл дистиллированной водой, приливают 2 мл раствора родамина «C», перемешивают и через 10 — 15 мин. измеряют оптическую плотность раствора при 610 нм в кювете с толщиной слоя 1 см по сравнению с контролем (раствор N 1 по таблице).
Строят градуировочный график зависимости оптической плотности от содержания цинка в градуировочном растворе (мкг), проверку которого проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой помещают в стакан и дважды промывают горячим раствором соляной кислоты 1:1 (по 5 мл). Промывные жидкости объединяют и выпаривают досуха в фарфоровой чашке на водяной бане. Сухой остаток растворяют в 10 мл дистиллированной воды. Аликвотную часть раствора (1 — 5 мл) помещают в пробирку и устанавливают pH раствора 3 — 4, добавляя 0,1 н. серную кислоту по каплям. Далее анализ идет аналогично приготовлению градуировочных растворов.
Оптическую плотность исследуемого раствора измеряют по сравнению с контролем, который готовят одновременно и аналогично пробам.
Содержание цинка в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию цинка в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета цинка на оксид цинка — 1,24.
Измерение концентрации молибдена
Характеристика метода
Определение основано на образовании оранжево-красного комплекса молибдена с роданидом аммония в присутствии восстановителя.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания молибдена в объеме анализируемого раствора 1 мкг.
Нижний предел измерения молибдена в воздухе 1 мг/куб. м (при отборе 25 л).
Диапазон измеряемых концентраций в воздухе от 1 мг/куб. м до 10 мг/куб. м.
Определению не мешают хром и никель в соотношении 40:1, ванадий и вольфрам — 70:1, железо в количестве до 1 мг, марганец, кобальт.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 2,5 часа, включая отбор пробы 2 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Муфельная печь, МП-2УМ.
Тигли фарфоровые, ГОСТ 9147-80С.
Щипцы тигельные.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25 и 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Пробирки колориметрические с притертыми пробками, ГОСТ 10515-75.
Стаканы химические, ГОСТ 25336-82Е.
Реактивы, растворы, материалы
Аммоний молибденовокислый (молибдат аммония), 4-водный ГОСТ 3765-78, хч.
Кислота азотная, ГОСТ 4461-77, чда, разбавленная 1:1.
Кислота серная, ГОСТ 4204-77, хч, 10% и разбавленная 1:1.
Аммоний роданистый (роданид аммония), СТ СЭВ 22-75, хч, 10% раствор.
Кислота аскорбиновая, ГОСТ 4815-76, хч, 5% раствор (свежеприготовленный).
Стандартный раствор N 1, содержащий 1 мг/мл молибдена, готовят растворением 0,1840 г молибдата аммония в 100 мл 10% раствора серной кислоты. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор N 2, содержащий 1 мкг/мл молибдена, готовят разбавлением раствора N 1 раствором серной кислоты в 100 раз, свежеприготовленный.
Фильтры типа АФА, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, укрепленный в фильтродержателе.
Пробы хранятся в течение месяца. Для измерения 1/2 ПДКм.р. молибдена следует отобрать 25 л воздуха.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 30 мин.) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МОЛИБДЕНА
| N стандарта | Стандартный раствор N 2, мл | Раствор серной кислоты, 10%, мл | Содержание молибдена в градуировочном растворе, мкг |
| 1 | 0 | 4,0 | 0 |
| 2 | 0,1 | 3,9 | 1,0 |
| 3 | 0,2 | 3,8 | 2,0 |
| 4 | 0,4 | 3,6 | 4,0 |
| 5 | 0,6 | 3,4 | 6,0 |
| 6 | 0,8 | 3,2 | 8,0 |
| 7 | 1,0 | 3,0 | 10,0 |
Во все пробирки прибавляют по 0,5 мл раствора роданида аммония и по 0,5 мл раствора аскорбиновой кислоты. Растворы перемешивают и через 30 минут фотометрируют при длине волны 480 нм в кюветах с толщиной слоя 1 см по сравнению с контролем (раствор N 1 шкалы стандартов).
Строят градуировочный график зависимости оптической плотности от содержания молибдена (мкг), проверку которого проводят 1 раз в квартал или при использовании новой партии реактивов.
Проведение измерения
Фильтр помещают в тигель и озоляют в муфельной печи при температуре 500 град. C. По охлаждении в тигель вносят 2 мл раствора серной кислоты 1:1 и 1 мл разбавленной кислоты. Содержимое тигля осторожно выпаривают на плитке до появления белых паров серного ангидрида. По охлаждении раствор осторожно разбавляют водой и количественно переносят в мерную колбу вместимостью 25 мл. Тигель промывают водой, сливая промывные воды в колбу. Доводят объем раствора в колбе до метки водой.
1 мл раствора пробы вносят в пробирку и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание молибдена в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию молибдена в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации кобальта и оксида кобальта
Характеристика метода
Определение основано на реакции иона кобальта с нитрозо-P-солью с образованием окрашенного в оранжево-розовый цвет соединения.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания кобальта в объеме анализируемого раствора 0,5 мкг.
Нижний предел измерения кобальта в воздухе 0,1 мг/куб. м (при отборе 25 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,1 до 10,0 мг/куб. м.
Определению не мешают железо в соотношении 500:1, медь — 100:1, никель — 50:1.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 2,5 часа, включая отбор проб 5 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Тигли фарфоровые, ГОСТ 9147-80Б.
Муфельная печь, МП-2УМ.
Щипцы тигельные.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 0,2, 1, 2 и 5 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 и 50 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Стаканы химические, ГОСТ 25336-82Е.
Реактивы, растворы, материалы
Кобальт двухлористый (хлорид кобальта), 6-водный, ГОСТ 4525-77, чда.
Кислота соляная, ГОСТ 3118-77, хч, концентрированная и 5% раствор.
Нитрозо-P-соль, ГОСТ 10553-75, чда, 0,1% раствор.
Кислота азотная, ГОСТ 4461-77, хч, концентрированная и разбавленная 1:1.
Натрий уксуснокислый (ацетат натрия), 3-водный, ГОСТ 199-78, хч, 50% и 5% растворы, свежеприготовленные.
Стандартный раствор N 1 с концентрацией 100 мкг/мл кобальта готовят растворением 0,0403 г хлорида кобальта, перекристаллизованного и высушенного на воздухе, в 100 мл дистиллированной воды в мерной колбе. Раствор устойчив в течение месяца.
Стандартный раствор N 2 с концентрацией 10 мкг/мл кобальта готовят разбавлением раствора N 1 дистиллированной водой в 10 раз.
Раствор устойчив в течение суток.
Фильтры АФА-ХА-20, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА-ХА-20. Пробы хранятся в течение месяца.
Для измерения 1/2 ПДК достаточно отобрать 25 л воздуха.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 30 минут) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ КОБАЛЬТА
| N стандарта | Стандартный раствор N 2, мл | Раствор ацетата натрия, 5%, мл | Содержание кобальта в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,05 | 4,95 | 0,5 |
| 3 | 0,1 | 4,9 | 1,0 |
| 4 | 0,2 | 4,8 | 2,0 |
| 5 | 0,4 | 4,6 | 4,0 |
| 6 | 0,6 | 4,4 | 6,0 |
| 7 | 0,8 | 4,2 | 8,0 |
| 8 | 1,0 | 4,0 | 10,0 |
Во все пробирки прибавляют по 1 мл раствора нитрозо-P-соли, 2 мл 50% раствора ацетата натрия и погружают их в кипящую водяную баню на 3 мин. По охлаждении приливают по 2 мл азотной кислоты (1:1). После прибавления каждого реактива растворы взбалтывают. Через 15 — 20 минут измеряют оптическую плотность растворов при длине волны 420 нм в кюветах с толщиной слоя 1 см по сравнению с контролем (раствор N 1 по таблице).
Строят градуировочный график зависимости оптической плотности градуировочных растворов от содержания кобальта (мкг), проверку которого производят один раз в квартал или в случае использования новых реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в тигель и озоляют при 500 град. C. В тигель наливают 5 мл концентрированной соляной кислоты, раствор доводят до кипения и переносят в фарфоровую чашку. Тигель промывают 5 — 10 мл соляной кислоты и 10 — 15 мл воды, каждый раз перенося смывы в ту же чашку. Раствор выпаривают досуха. К сухому остатку приливают 5 — 7 мл 5% раствора ацетата натрия, тщательно растирают остаток стеклянной палочкой и раствор фильтруют через фильтр «синяя лента» диаметром 3 см в мерную колбу вместимостью 25 мл. Чашку еще 3 — 4 раза обрабатывают раствором ацетата натрия, собирая смывы в ту же колбу. Раствор в колбе доводят до метки раствором ацетата натрия и перемешивают.
1 — 5 мл полученного раствора пробы вносят в колориметрические пробирки и далее анализируют и фотометрируют аналогично градуировочным растворам.
Содержание кобальта в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию кобальта в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации оксидов ванадия
Характеристика метода
Определение основано на реакции окисления оксидов ванадия до пятивалентного состояния и последующем фотометрическом измерении окрашенного в пурпурный цвет комплексного соединения с 4-(2-пиридилазо) резорцином в буферном растворе с pH 6,0.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения концентрации оксида ванадия (V) в фотометрируемом объеме раствора составляет 2,5 мкг.
Нижний предел измерения оксида ванадия (V) в воздухе составляет 0,05 мг/куб. м (при отборе 90 л воздуха).
Диапазон измеряемых концентраций оксида ванадия (V) в воздухе от 0,05 до 1,4 мг/куб. м.
Измерению ванадия не мешают 10-кратные избытки титана, кальция, тантала, хрома (III и VI). Мешающие измерению железо, кобальт, никель, кадмий, медь, цинк, свинец, марганец, молибден, вольфрам отделяются от ванадия в ходе подготовки пробы к анализу.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 6 ч, включая отбор пробы воздуха.
Приборы, аппаратура, посуда
Спектрофотометр или фотоэлектроколориметр.
pH-метр pH-121 или pH-340.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Тигли кварцевые, ГОСТ 19908-80, или платиновые, ГОСТ 6563-75.
Щипцы тигельные.
Электроплитка.
Воронки химические, ГОСТ 25336-82.
Колбы мерные, ГОСТ 1770-74, вместимостью 25 и 1000 мл.
Пипетки, ГОСТ 20292-74, вместимостью 1, 2, 5 и 10 мл.
Реактивы, растворы и материалы
Аммоний ванадиевокислый мета (NH4VO3), ГОСТ 9336-75, чда.
Натрия гидроксид, ГОСТ 4328-77, хч, 4% раствор.
Соляная кислота, ГОСТ 3118-77, хч, разбавленная 1:1 (по объему).
Аммоний уксуснокислый, ГОСТ 3117-78, чда.
Уксусная кислота, ГОСТ 61-75, хч, 24% раствор.
Азотная кислота, ГОСТ 4461-77, хч, 50% раствор (по объему).
Калий-натрий углекислый, ГОСТ 4332-76, хч.
4-(2-пиридилазо) резорцин (ПАР), МРТУ 6-09-2882-66, 0,03% водный раствор.
Этиловый спирт, ГОСТ 5963-67.
Буферный раствор с pH 6,0 готовят путем растворения в воде 77 г уксуснокислого аммония, добавления 10 мл раствора уксусной кислоты и доведения объема в мерной колбе до 1 л дистиллированной водой (pH раствора следует проверить на pH-метре и в случае необходимости добавить кислоты или щелочи).
Стандартный раствор N 1 с концентрацией оксида ванадия (V) 1 мг/мл готовят растворением 1,286 г ванадия аммония в 1 л воды. Раствор устойчив более года.
Стандартный раствор N 2 с концентрацией оксида ванадия (V) 5,0 мкг/мл (применяют свежеприготовленным) готовят путем соответствующего разбавления водой стандартного раствора N 1.
Фильтры обеззоленные «белая лента», ТУ 6-09-1678-77.
Индикаторная бумага универсальная, ТУ 6-09-1181-76.
Фильтры АФА-ХА-20, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА-ХА-20, помещенный в фильтродержатель. Для измерения 1/2 ПДК (в расчете на дым ванадия (V) оксида) следует отобрать 90 л воздуха. Отобранные пробы устойчивы в течение месяца.
Подготовка к измерению
Градуировочные растворы оксида ванадия (V) (устойчивы в течение суток) готовят в мерных колбах вместимостью 25 мл. В колбы вносят стандартный раствор N 2 в количествах, указанных в таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ
| N раствора | Стандартный раствор N 2, мл | Содержание оксида ванадия (V) в 25 мл градуировочного раствора, мкг |
| 1 | 0 | 0,0 |
| 2 | 0,5 | 2,5 |
| 3 | 1,0 | 5,0 |
| 4 | 2,0 | 10,0 |
| 5 | 4,0 | 20,0 |
| 6 | 8,0 | 40,0 |
| 7 | 10,0 | 50,0 |
| 8 | 15,0 | 75,0 |
Во все колбы добавляют по 5 мл буферного раствора, по 2 мл раствора ПАР и доводят объемы до метки дистиллированной водой. Растворы перемешивают и через 10 мин. измеряют оптическую плотность при длине волны 540 нм. Измерение проводят в кюветах с толщиной слоя 1 см по отношению к контрольному раствору.
Строят градуировочный график зависимости оптической плотности от содержания оксида ванадия (V) в растворах (мкг), проверку которого проводят 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в тигель, добавляют 2 мл 50-процентного раствора азотной кислоты, выпаривают досуха на плитке. Затем пробу озоляют в муфельной печи (предварительно закрыв тигель крышкой) в течение 1 часа при постепенном повышении температуры до 500 град. C. Зольный остаток смешивают с 0,2 — 0,3 г калия-натрия углекислого, помещают для сплавления в муфельную печь, температуру которой повышают до 750 — 800 град. C и выдерживают 10 мин. Плав растворяют в воде при кипячении на плитке, содержимое количественно переносят в мерную колбу вместимостью 25 мл, добавляют 1 — 2 капли этилового спирта (для восстановления оксидов марганца, если они присутствуют), дают осадку гидроксидов скоагулироваться, раствор охлаждают и объем доводят водой до метки. Раствор пробы фильтруют через беззольный фильтр (отделяют гидроксиды железа, титана, кальция и др.) и в фильтрате определяют концентрацию оксида ванадия (V).
Для этого отбирают аликвоту фильтрата объемом 15 мл, нейтрализуют соляной кислотой по универсальной индикаторной бумаге до pH 5 — 6, добавляют 5 мл буферного раствора, 2 мл раствора ПАР и объем доводят водой до 25 мл. Оптическую плотность измеряют аналогично градуировочным растворам.
Содержание оксида ванадия (V) в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию оксида ванадия (V) в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентраций вольфрама
Характеристика метода
Определение основано на реакции взаимодействия вольфрамат-иона с роданидом аммония с образованием комплексного соединения, окрашенного в желтый цвет.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания вольфрама в объеме анализируемого раствора 2 мкг.
Нижний предел измерения вольфрама в воздухе 1,3 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 1,3 до 62 мг/куб. м. Определению не мешают титан, кобальт, железо, марганец, хром и ванадий.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 1,5 часа, включая отбор пробы 2 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Тигли фарфоровые, ГОСТ 9147-80Е.
Щипцы тигельные.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25 и 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 0,2, 1, 2 и 5 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Стаканы химические, ГОСТ 25336-82, вместимостью 50 и 100 мл.
Реактивы, растворы, материалы
Натрий вольфрамовокислый (вольфрамат натрия), 2-водный, ГОСТ 18289-78, чда.
Вольфрам (VI) оксид (вольфрамовый ангидрид), ТУ 6-09-4236-76, чда.
Натрия гидроксид, ГОСТ 4328-77, хч, 10% и 2% растворы.
Олово двухлористое, ГОСТ 36-78, чда, 12% раствор в концентрированной соляной кислоте (растворяют при нагревании).
Аммоний роданистый (роданид аммония), СТ СЭВ 222-75, хч, или калий роданистый (роданид калия), ГОСТ 4139-75, хч, 25% раствор.
Стандартный раствор вольфрама N 1 с концентрацией 1 мг/мл готовят путем растворения 0,1794 г вольфрамата натрия в 2% растворе гидроксида натрия в мерной колбе вместимостью 100 мл либо растворением 0,1261 г оксида вольфрама (VI) при нагревании в 20 мл 10% раствора гидроксида натрия, количественным переведением полученного раствора в мерную колбу вместимостью 100 мл и доведением объема до метки дистиллированной водой. Раствор устойчив в течение 3 месяцев.
Стандартные растворы вольфрама N 2 и N 3 с концентрацией 100 и 10 мкг/мл готовят путем соответствующего разбавления раствора N 1 2% раствором гидроксида натрия в мерных колбах. Раствор N 2 устойчив в течение месяца, N 3 применяют свежеприготовленным.
Фильтры типа АФА, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр типа АФА, помещенный в фильтродержатель.
Для измерения 1/2 ПДК достаточно отобрать 20 л воздуха.
Пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2 час.) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ВОЛЬФРАМА
| N стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Гидроксид натрия, 2%, раствор, мл | Содержание вольфрама в градуировочном растворе, мкг |
| 1 | — | — | 2,0 | 0 |
| 2 | 0,2 | — | 1,8 | 2,0 |
| 3 | 0,5 | — | 1,5 | 5,0 |
| 4 | 1,0 | — | 1,0 | 10,0 |
| 5 | 2,0 | — | 0,0 | 20,0 |
| 6 | — | 0,4 | 1,6 | 40,0 |
| 7 | — | 0,6 | 1,4 | 60,0 |
| 8 | — | 0,8 | 1,2 | 80,0 |
| 9 | — | 1,0 | 1,0 | 100,0 |
Во все пробирки шкалы прибавляют по 0,2 мл раствора роданида аммония, взбалтывают, приливают по 2,8 мл раствора хлорида олова и снова взбалтывают. Через 1 час измеряют величину оптической плотности растворов при длине волны 420 нм в кюветах с толщиной слоя 1 см по сравнению с контрольным раствором (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности от содержания вольфрама в градуировочном растворе (мкг), проверка которого производится 1 раз в квартал или в случае использования новых реактивов.
Проведение измерения
Фильтр с отобранной пробой помещают в тигель и озоляют в муфельной печи при температуре 500 град. C, зольный остаток выдерживают в печи при температуре 500 — 600 град. C около 20 минут для получения трехокиси вольфрама. По охлаждении остаток в тигле обрабатывают 2 мл 10% раствора гидроксида натрия и нагревают до полного растворения остатка. Раствор переводят в мерную колбу вместимостью 25 мл, а тигель промывают водой, сливая ее в ту же колбу. Полученный раствор доводят до метки водой. Если раствор мутный, его центрифугируют или фильтруют.
2 мл пробы вносят в колориметрическую пробирку и далее анализируют и фотометрируют аналогично градуировочным растворам.
Содержание вольфрама в анализируемом объеме раствора пробы (мкг) находят по градуировочному графику.
Концентрацию вольфрама в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации циркония и оксида циркония (IV)
Характеристика метода
Определение основано на реакции циркония-иона с пирокатехиновым фиолетовым с образованием окрашенного соединения.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания циркония в объеме анализируемого раствора 5 мкг.
Нижний предел измерения в воздухе 0,5 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,5 мг/куб. м до 20 мг/куб. м.
Определению не мешают железо, алюминий, ванадий и титан до содержания 100 мкг.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 3 часа, включая отбор проб 2 минуты.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня водяная.
Печь муфельная, МП-2УМ.
Щипцы тигельные с платиновыми наконечниками.
Центрифуга.
Тигли платиновые, ГОСТ 6563-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50 и 100 мл.
Пробирки центрифужные, ГОСТ 25336-82Е, вместимостью 15 мл.
Ступка фарфоровая с пестиком, ГОСТ 9147-80Е.
Чашки фарфоровые, ГОСТ 9147-80Е.
Реактивы, растворы, материалы
Цирконий хлорокись, 8-водная, МРТУ 6-09-2901-76, хч.
Пирокатехиновый фиолетовый, ТУ 6-09-07-1087-78, чда, 0,1% раствор.
Соляная кислота, ГОСТ 3118-77, хч, 0,5 М и 2 растворы.
Аммоний уксуснокислый, ГОСТ 3117-78, хч.
Кислота уксусная ледяная, ГОСТ 61-75, хч.
Метиловый оранжевый, ГОСТ 10861-64, 0,05% раствор.
Натрий углекислый, безводный, ГОСТ 83-79, хч.
Натрий тетраборнокислый, 10-водный, ГОСТ 4199-76, хч.
Магний хлористый, безводный, МРТУ 6-09-3237-66.
Плавень: тщательно растирают в фарфоровой ступке 3 части карбоната натрия и 2 части прокаленного натрия тетрабората. К 100 г полученной смеси прибавляют 1 г безводного хлорида магния и снова растирают в однородную массу. Хранят в склянке с притертой пробкой.
Буферный раствор. 27 г ацетата аммония растворяют в 900 мл воды в мерной колбе вместимостью 1 л, прибавляют по каплям уксусную кислоту до pH 5,3 и доводят объем до метки водой.
Трилон Б, ГОСТ 10652-73, 5% раствор.
Стандартный раствор циркония N 1 с концентрацией 100 мкг/мл готовят путем растворения 0,3530 г хлорида циркония в 0,5 М растворе соляной кислоты в мерной колбе вместимостью 1 л. Раствор устойчив в течение 1 месяца.
Стандартный раствор циркония N 2 с концентрацией 10 мкг/мл готовят разбавлением основного раствора 0,5 М раствором соляной кислоты в 10 раз. Раствор устойчив в течение 2 дней.
Фильтры обеззоленные «синяя лента», ТУ 6-09-1678-77.
Фильтры типа АФА, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр, АФА, помещенный в фильтродержатель.
Для определения 1/2 ПДК циркония следует отобрать 20 л воздуха. Проба сохраняется в течение 1 месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят в мерных колбах вместимостью 25 мл согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЦИРКОНИЯ
| N стандарта | Стандартный раствор N 2, мл | Соляная кислота, мл | Содержание циркония в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,5 | 4,5 | 5,0 |
| 3 | 1,0 | 4,0 | 10,0 |
| 4 | 2,0 | 3,0 | 20,0 |
| 5 | 3,0 | 2,0 | 30,0 |
| 6 | 4,0 | 1,0 | 40,0 |
Во все колбы шкалы приливают по 2 мл раствора Трилона Б, 1 каплю метилового оранжевого и осторожно по каплям нейтрализуют раствором аммиака до перехода окраски в желтый цвет. Затем прибавляют по каплям 2 М раствор соляной кислоты до перехода окраски в оранжевый цвет, 8 — 10 мл буферного раствора, 0,7 мл раствора пирокатехинового фиолетового, доводят раствор до метки буферным раствором и перемешивают. Через 30 минут измеряют оптическую плотность раствора при длине волны 530 нм в кюветах с толщиной слоя 0,5 см относительно контрольного раствора (раствор 1 по таблице).
Строят градуировочный график зависимости оптической плотности растворов от содержания циркония (мкг), проверку которого проводят 1 раз в квартал или при использовании новой партии реактивов.
Проведение измерения
Фильтр с пробой помещают в платиновый тигель и осторожно, избегая воспламенения, озоляют на газовой горелке или в муфельной печи. К остатку добавляют 2 г плавня и помещают тигель в переднюю часть нагретой муфельной печи. После прекращения вспенивания плавня тигель продвигают на середину печи и выдерживают до полной прозрачности плава. При температуре около 900 град. C продолжают сплавление в течение 5 минут. По охлаждении в тигель наливают 10 мл воды и помещают на кипящую водяную баню.
Когда плав растворится, жидкость с осадком переносят в центрифужную пробирку, тигель промывают 2 мл воды, сливая раствор в ту же пробирку. Раствор центрифугируют при 2000 об./мин. в течение 7 — 10 минут. Затем жидкость сливают с осадка, осадок промывают водой дважды по 5 мл, каждый раз центрифугируя взвесь. Раствор отбрасывают и растворяют осадок в 10 мл 2 М раствора соляной кислоты при нагревании на кипящей водяной бане. Раствор перемешивают стеклянной палочкой и охлаждают.
1 — 5 мл полученного раствора вносят в мерную колбу вместимостью 25 мл, приливают 2 М раствор соляной кислоты до объема 5 мл и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание циркония в анализируемом объеме раствора пробы (мкг) проводят по градуировочному графику.
Концентрацию циркония в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации алюминия и оксида алюминия
Характеристика метода
Определение основано на реакции взаимодействия иона алюминия с хромазуролом S с образованием соединения, окрашенного в фиолетово-синий цвет.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания алюминия — 0,3 мкг в анализируемом объеме раствора.
Нижний предел измерения алюминия в воздухе — 0,4 мг/куб. м, оксида алюминия 7,5 мг/куб. м (при отборе 80 л воздуха).
Диапазон измеряемых концентраций алюминия в воздухе от 0,4 до 30 мг/куб. м.
Определению не мешают кальций, магний, свинец, цинк, 25-кратные количества железа, никеля, меди, марганца, 2-кратные количества хрома и титана. Мешающие определению ионы фтора отделяют в ходе обработки пробы. Мешающее влияние одновалентных катионов устраняют путем разбавления пробы.
Суммарная погрешность измерения не превышает +/- 20%. Время выполнения измерения 5 — 6 часов, включая отбор пробы 8 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56 или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Тигли платиновые, ГОСТ 6563-75.
Щипцы тигельные с платиновыми наконечниками.
Баня песчаная.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5, 10 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 50, 100, 500 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 25, 50, 100 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 мл.
Реактивы, растворы, материалы
Квасцы алюмокалиевые (калий-алюминий сернокислый 12-водный), ГОСТ 4329-77, чда.
Калия пиросульфат (калий сернокислый пиро), ГОСТ 7172-76, чда.
Кислота серная, ГОСТ 4204-77, хч, концентрированная и разбавленная (1:1).
Кислота фтористоводородная, ГОСТ 10484-73, чда, 45%.
Кислота аскорбиновая, ГОСТ 4815-74, хч, 1% водный раствор, свежеприготовленный.
Кислота винная, ГОСТ 5817-77, чда, 0,05% водный раствор.
Тиомочевина, ГОСТ 6344-73, хч, 5% раствор.
Кислота уксусная, ГОСТ 61-75, хч, 0,2 М раствор.
Аммиак водный, ГОСТ 3760-64, хч, 0,2 М раствор.
Буферный раствор с pH 7,5. Смешивают 49,6 мл 0,2 М раствора уксусной кислоты и 50,4 мл 0,2 М раствора аммиака. Проверка pH раствора на pH-метре обязательна. Буферный раствор устойчив в течение недели.
Хромазурол S, ТУ 6-09-4513-77, чда, 0,1% раствор. Раствор устойчив в течение месяца.
Раствор плава. Пиросульфат калия в количестве 0,5 г помещают в платиновый тигель и плавят при температуре 650 — 700 град. C. Плав растворяют в горячей воде и переводят в мерную колбу вместимостью 500 мл.
Стандартный раствор алюминия N 1 с концентрацией 100 мкг/мл готовят растворением 0,1758 г алюмокалиевых квасцов в 50 мл теплой воды с добавлением 1 мл концентрированной азотной кислоты в мерной колбе вместимостью 100 мл. После охлаждения раствор доводят до метки дистиллированной водой. Раствор устойчив в течение месяца.
Стандартные растворы N 2 и N 3 с концентрацией алюминия 10 и 1,0 мкг/мл готовят соответствующим разбавлением основного раствора раствором плава. Растворы устойчивы в течение одного дня.
Фильтры АФА-ХА или ХП, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, помещенный в фильтродержатель. Для измерения 1/2 ПДК следует отобрать 80 л воздуха. Пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 20 мин.) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ АЛЮМИНИЯ
| N стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Раствор плава, мл | Содержание алюминия в градуировочном растворе, мкг |
| 1 | 0 | — | 5,0 | 0 |
| 2 | 0,3 | — | 4,7 | 0,3 |
| 3 | 0,5 | — | 4,5 | 0,5 |
| 4 | 1,0 | — | 4,0 | 1,0 |
| 5 | — | 0,2 | 4,8 | 2,0 |
| 6 | — | 0,3 | 4,7 | 3,0 |
| 7 | — | 0,4 | 4,6 | 4,0 |
| 8 | — | 0,5 | 4,5 | 5,0 |
Во все пробирки вносят по 0,3 мл 1% раствора аскорбиновой кислоты, 0,25 мл 5% раствора тиомочевины, 0,2 мл 0,05% раствора винной кислоты, 4,0 мл буферного раствора и 0,3 мл 0,1-процентного раствора хромазурола S. После добавления каждого реактива содержимое пробирок перемешивают.
Через 15 минут измеряют величину оптической плотности растворов при длине волны 530 нм в кюветах с толщиной слоя 1 см по отношению к контрольному раствору (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности от содержания алюминия в градуировочном растворе (мкг), проверка которого проводится 1 раз в квартал в случае использования новой партии реактивов.
Проведение измерения
Фильтр АФА-ХА с пробой помещают в платиновый тигель, вносят 3 — 4 капли воды, 6 — 8 капель серной кислоты (1:1), 2 мл фтористоводородной кислоты и выпаривают до удаления паров на песчаной бане. При отборе проб на фильтры АФА-ХП кислотной обработке предшествует операция озоления фильтра при температуре 500 град. C. Затем тигель помещают в муфельную печь и прокаливают при температуре 900 — 1000 град. C в течение 5 — 10 мин. После охлаждения в тигель вносят 0,5 г пиросульфата калия, хорошо перемешивают и сплавляют, постепенно повышая температуру до 650 — 700 град. C, выдерживают при данной температуре 10 минут, затем охлаждают. Плав растворяют в горячей воде, переводят в мерную колбу вместимостью 500 мл и после охлаждения раствор в колбе доводят до метки водой.
В колориметрическую пробирку вносят 1 — 5 мл пробы и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание алюминия в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию алюминия в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета алюминия на оксид алюминия — 1,88.
Измерение концентрации магния и оксида магния
Характеристика метода
Определение основано на реакции образования окрашенного комплексного соединения магния с арсеназо-1.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения магния в анализируемом объеме раствора 1 мкг.
Нижний предел измерения в воздухе 1 мг/куб. м (при отборе 25 л воздуха).
Диапазон измеряемых концентраций в воздухе от 1 мг/куб. м до 20 мг/куб. м.
Определению магния не мешают алюминий, железо, медь, вольфрам.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 2 часа, включая отбор пробы 2,5 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74, 1-й класс.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Тигли платиновые, ГОСТ 6563-75.
Щипцы тигельные.
Баня водяная.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 100 и 1000 мл.
Пробирки колориметрические ГОСТ 10515-75.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 мл.
Реактивы, растворы, материалы
Магний сернокислый, 7-водный, ГОСТ 4523-77, хч.
Спирт этиловый, ГОСТ 18300-72, хч.
Арсеназо-1, МРТУ 6-09-4729-79, чда, перекристаллизованный из смеси этилового спирта с водой в соотношении 1:1 с последующим промыванием полученного осадка этиловым спиртом и высушиванием его при комнатной температуре, 0,06% раствор.
Аммиак водный, ГОСТ 3760-79, чда, 0,2 М раствор (15,1 мл 25% раствора аммиака растворяют в 1000 мл дистиллированной воды).
Уксусная кислота ледяная, ГОСТ 61-75, хч, 0,2 М раствор (11,62 мл ледяной уксусной кислоты растворяют в 1000 мл дистиллированной воды); 10% раствор.
Аммиачно-ацетатный буферный раствор с pH 10,4 — 11,0. 976 мл 0,2 М раствора аммиака смешивают с 24 мл 0,2 М раствора уксусной кислоты. Раствор устойчив в течение месяца.
Триэтаноламин, ТУ 6-09-2448-72, ч, 5% водный раствор.
Калий пиросернокислый (пиросульфат), ГОСТ 7172-76, ч.
Стандартный раствор магния N 1 с концентрацией 1 мг/мл магния готовят растворением 10,1386 г сернокислого магния в дистиллированной воде в мерной колбе вместимостью 1000 мл. Объем доводят до метки дистиллированной водой. Раствор устойчив в течение месяца.
Стандартный раствор магния N 2 с концентрацией 10 мкг/мл готовят соответствующим разведением стандартного раствора N 1 дистиллированной водой в день анализа.
Фильтры типа АФА, ТУ 95-843-80.
Отбор пробы воздуха
Воздух с объемным расходом 5 — 10 л/мин. аспирируют через фильтр АФА.
Для измерения 1/2 ПДК магния достаточно отобрать 25 л воздуха.
Пробы хранятся в течение 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 30 мин.) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МАГНИЯ
| N стандарта | Стандартный раствор магния N 2, мл | Дистиллированная вода, мл | Содержание магния в градуировочном растворе, мкг |
| 1 | 0 | 4,0 | 0 |
| 2 | 0,1 | 3,9 | 1,0 |
| 3 | 0,2 | 3,8 | 2,0 |
| 4 | 0,3 | 3,7 | 3,0 |
| 5 | 0,4 | 3,6 | 4,0 |
| 6 | 0,5 | 3,5 | 5,0 |
| 7 | 0,6 | 3,4 | 6,0 |
| 8 | 0,7 | 3,3 | 7,0 |
| 9 | 0,9 | 3,1 | 9,0 |
| 10 | 1,0 | 3,0 | 10,0 |
Во все пробирки шкалы добавляют по 1 мл раствора триэтаноламина, по 3 мл аммиачно-ацетатного буферного раствора (pH 10,4 — 11), по 2 мл 0,06% раствора арсеназо-1. После добавления каждого реактива содержимое пробирок перемешивают. Объем растворов доводят дистиллированной водой до 10 мл, перемешивают и через 15 мин. фотометрируют. Измерение оптической плотности проводят в кюветах с толщиной поглощающего слоя 2 см при 572 — 592 нм.
Строят градуировочный график зависимости оптической плотности от содержания магния в градуировочных растворах (мкг).
Проверка градуировочного графика проводится 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с пробой помещают в платиновый тигель и озоляют при 500 град. C в течение 30 минут. После охлаждения в тигель вносят 0,5 г пиросульфата калия, хорошо перемешивают и сплавляют, постепенно повышая температуру до 600 град. C, в течение 30 мин. По охлаждении плав растворяют в горячей дистиллированной воде, переносят раствор в мерную колбу вместимостью 25 мл и доводят раствор до метки водой.
0,5 — 1 мл пробы вносят в колориметрическую пробирку, доводят объем до 4,0 мл водой, и далее анализ идет аналогично приготовлению градуировочных растворов.
Оптическую плотность полученных растворов измеряют аналогично градуировочным растворам по сравнению с контролем, который готовят одновременно и аналогично пробе.
Содержание магния в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию магния в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета магния на оксид магния 1,66.
Измерение концентрации свинца
Характеристика метода
Определение основано на взаимодействии иона свинца с сульфарсазеном с образованием комплексного соединения, окрашенного в желто-оранжевый цвет.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения в анализируемом объеме раствора 1 мкг.
Нижний предел намерения в воздухе 0,005 мг/куб. м (при отборе 400 л воздуха).
Диапазон измеряемых концентраций свинца в воздухе от 0,005 до 0,12 мг/куб. м.
Определению свинца не мешают цинк, медь, железо.
Суммарная погрешность намерения не превышает +/- 25%.
Время выполнения определения 1,5 часа, включая отбор пробы 20 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр марки ФЭК-56М или другой системы, ГОСТ 15150-74.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня песчаная.
Печь муфельная, МП-2УМ.
Пробирки колориметрические, ГОСТ 10515-75, вместимостью 10 мл, с пришлифованными пробками.
Пробирки центрифужные, ГОСТ 25336-82Е.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25 и 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 и 100 мл.
Реактивы, растворы, материалы
Свинец азотнокислый, ГОСТ 4236-77, хч, перекристаллизованный и высушенный до постоянного веса.
Аммоний уксуснокислый (ацетат аммония), ГОСТ 3117-78, хч, 3% раствор.
Натрий тетраборнокислый, 10-водный (бура), ГОСТ 4199-76, хч, 0,05 М раствор, готовят растворением 9,51 г буры в 500 мл воды.
Тиомочевина, ГОСТ 6344-73, хч, 10% раствор.
Сульфарсазен, ТУ 6-09-4681-78, чда, 0,025% в 0,05 М растворе буры. Раствор устойчив в течение 2 недель.
Калий железистосинеродистый (желтая кровяная соль), ГОСТ 4207-75, хч, 1% раствор.
Азотная кислота, ГОСТ 4461-77, хч, раствор 1:3.
Стандартный раствор свинца N 1 с концентрацией 1 мг/мл готовят растворением навески азотнокислого свинца 0,1598 г в воде в мерной колбе вместимостью 100 мл. Раствор устойчив в течение месяца.
Стандартный раствор свинца N 2 с концентрацией 100 мкг/мл готовят соответствующим разбавлением стандартного раствора N 1 3% раствором ацетата аммония. Раствор устойчив в течение недели.
Стандартный раствор N 3, содержащий 10 мкг/мл свинца, готовят разбавлением стандартного раствора N 2 раствором ацетата аммония. Применяют свежеприготовленным.
Фильтры АФА-ХП-20 или АФА-ВП-20, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 20 л/мин. аспирируют через фильтр АФА-ХП-20, закрепленный в фильтродержатель. Для измерения 1/2 ПДК свинца следует отобрать 400 л воздуха. Пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 5 часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ СВИНЦА
| N стандарта | Стандартный раствор магния N 3, мл | Раствор ацетата аммония 3%-ный, мл | Содержание свинца в градуировочном растворе, мкг |
| 1 | 0 | 2,5 | 0 |
| 2 | 0,1 | 2,4 | 1,0 |
| 3 | 0,2 | 2,3 | 2,0 |
| 4 | 0,4 | 2,1 | 4,0 |
| 5 | 0,6 | 1,9 | 6,0 |
| 6 | 0,8 | 1,7 | 8,0 |
| 7 | 1,0 | 1,5 | 10,0 |
Во все пробирки шкалы добавляют по 0,2 мл 10% раствора тиомочевины (для связывания ионов меди), по 0,1 мл 1% раствора желтой кровяной соли (для связывания ионов цинка), по 0,05 М растворов буры (в качестве буферного раствора) и по 0,5 мл 0,025-процентного раствора сульфасазена. После каждого раствора содержимое пробирок перемешивают. Выдерживают 30 минут, затем фотометрируют.
Измерение оптической плотности проводят при длине волны 540 нм по сравнению с контролем (раствор N 1 по таблице) в кюветах с толщиной слоя 1 см.
Строят градуировочный график зависимости оптической плотности растворов от содержания свинца (мкг), проверку которого проводят 1 раз в квартал или при использовании новой партии реактивов.
Проведение измерения
Фильтр с пробой переносят в химический стакан, заливают 10 мл раствора азотной кислоты (1:3) и нагревают при перемешивании стеклянной палочкой на кипящей водяной бане в течение 7 — 10 минут. Фильтр отжимают стеклянной палочкой на стенке стакана, промывают дистиллированной водой дважды по 5 мл. Раствор упаривают досуха на водяной бане. В стакан добавляют 3 — 5 мл воды и вновь упаривают досуха.
В стакан приливают 5 мл 3% раствора ацетата аммония, тщательно растирая осадок палочкой. При необходимости раствор центрифугируют. 1 — 2,5 мл прозрачного раствора помещают в колориметрические пробирки и обрабатывают и фотометрируют аналогично градуировочным растворам. Измерение оптической плотности раствора пробы проводят относительно усредненного контрольного раствора, полученного путем обработки (аналогичной обработке пробы) не менее 3 — 4 чистых фильтров.
Содержание свинца в анализируемом растворе пробы (мкг) находят по градуировочному графику.
Концентрацию свинца в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации борной кислоты и борного ангидрида
Характеристика метода
Определение основано на реакции борат-ионов с 1,1-диантримидом в концентрированной серной кислоте с образованием окрашенного в голубой цвет комплексного соединения.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения концентрации борной кислоты в анализируемом объеме раствора 1 мкг.
Нижний предел измерения в воздухе 0,3 мг/куб. м (при отборе 40 л воздуха).
Диапазон измеряемых концентраций борной кислоты в воздухе от 0,3 мг/куб. м до 37,5 мг/куб. м.
Определению не мешают металлы. Фторид-ионы мешают при соотношении концентраций борат-ионов и фтор-ионов 1:2,5 и выше.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 1 час 20 минут, включая отбор пробы 4 минуты.
Приборы, аппаратура, посуда
Фотоэлектроколориметр ФЭК-56М или другой марки, ГОСТ 15150-74, 1-й класс точности.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл. Колбы мерные, ГОСТ 177-74, вместимостью 50, 100 и 500 мл.
Чашки фарфоровые, ГОСТ 9147-73, вместимостью 50 мл.
Баня водяная.
Реактивы, растворы, материалы
Борная кислота, ГОСТ 9658-75, хч.
Серная кислота концентрированная, ГОСТ 4204-77, хч.
Диантримид, ТУ 05-9/691-77, чда, 0,4% (об.) раствор в серной кислоте. Раствор устойчив в течение 1 месяца при хранении при +4 град. C. В день анализа готовят 0,02% раствор реактива, для чего 1 объем 0,4-процентного раствора диантримида смешивают с 20 объемами серной кислоты.
Натрия гидроксид, ГОСТ 4328-77, хч, 0,01 М раствор.
Стандартный раствор N 1 борной кислоты с концентрацией 50 мкг/мл готовят растворением 0,0500 г борной кислоты с концентрированной серной кислоте в мерной колбе вместимостью 100 мл. Навеску вносят в колбу, приливают 30 — 50 мл серной кислоты, закрывают пришлифованной пробкой и осторожно перемешивают при нагревании на водяной бане до полного растворения или выдерживают в течение суток.
Раствор в колбе, охлажденный до комнатной температуре, доводят до метки серной кислотой и перемешивают.
Раствор устойчив в течение 1 месяца при хранении в стеклянной посуде, не содержащей бора.
Стандартный раствор N 2, содержащий 5 мкг/мл борной кислоты, готовят разбавлением основного раствора серной кислоты в 10 раз. Раствор устойчив в течение 6 часов.
Фильтры типы АФА, ТУ 95-743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, помещенный в фильтродержатель. Для измерения 1/2 ПДК следует отобрать 40 л воздуха. Проба хранится в течение 1 месяца.
Подготовка к измерению
Градуировочные растворы готовят согласно таблице. Устойчивы в течение 2-х часов.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ БОРНОЙ КИСЛОТЫ
| N стандарта | Стандартный раствор N 1, мл | Стандартный раствор N 2, мл | Серная кислота, мл | Содержание борной кислоты, мкг |
| 1 | — | — | 2,0 | 0 |
| 2 | — | 0,2 | 1,8 | 1,0 |
| 3 | — | 0,4 | 1,6 | 2,0 |
| 4 | — | 0,6 | 1,4 | 3,0 |
| 5 | — | 1,0 | 1,0 | 5,0 |
| 6 | — | 2,0 | — | 10,0 |
| 7 | 0,4 | — | 1,6 | 20,0 |
| 8 | 0,6 | — | 1,4 | 30,0 |
В пробирки вносят по 3 мл 0,02% (об.) раствора диантримида в серной кислоте, осторожно перемешивают и содержимое нагревают на кипящей водяной бане в течение 30 минут. По охлаждении измеряют оптическую плотность растворов при 620 нм в кюветах с толщиной слоя 1 см относительно контрольного раствора (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности от содержания борной кислоты в распорах (мкг), проверку которого проводят 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с пробой помещают в фарфоровую чашку и обрабатывают 7 — 10 мл горячей (70 град. C) дистиллированной воды, перемешивая стеклянной палочкой в течение 3 минут. Фильтр отжимают на стенке чашки стеклянной палочкой, промывают 10 мл воды и отбрасывают.
В чашку приливают 2 мл 0,01 М раствора гидроксида натрия, помещают на кипящую водяную баню и выпаривают досуха. В чашку приливают 10 мл серной кислоты, перемешивают содержимое стеклянной палочкой и сливают раствор в мерную колбу вместимостью 25 мл. Промывают чашку серной кислотой, собирая смывы в ту же колбу. Объем раствора в колбе доводят до метки серной кислотой и перемешивают.
0,5 — 2,0 мл раствора пробы помещают в пробирку, доводят в случае необходимости объем раствора до 2,0 мл серной кислотой и приливают 3,0 мл раствора диантримида. Дальше анализ проводят аналогично градуировочным растворам.
Оптическую плотность раствора пробы измеряют аналогично градуировочным растворам по сравнению с контролем, который готовят одновременно и аналогично пробе.
Количественное определение борной кислоты в мкг в анализируемом объеме раствора проводят по градуировочному графику.
Расчет концентрации борной кислоты в воздухе (мг/куб. м) проводят по формуле 1 (Приложение 4.2).
При определении борного ангидрида полученное значение концентрации следует умножить на коэффициент 0,57.
Измерение концентрации диоксида кремния
Характеристика метода
Определение кристаллической и аморфной (из аэрозоля конденсации) форм диоксида кремния основано на переведении их в раствор, путем сплавления со смесью двууглекислого натрия и хлорида натрия и последующей реакции образования синего кремнемолибденового комплекса.
Отбор проб проводится с концентрированием на фильтр.
Нижний предел измерения содержания диоксида кремния в объеме анализируемого раствора 2 мкг.
Нижний предел измерения диоксида кремния в воздухе 0,5 мг/куб. м (при отборе 100 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,5 до 12,5 мг/куб. м.
Определению диоксида кремния не мешают соединения фосфора.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 7 — 8 часов, включая отбор проб 8 — 10 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр ФЭК-56М или другой марки, ГОСТ 15150-74, 1-й класс точности.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Тигли платиновые, ГОСТ 6563-75, с крышками.
Печь муфельная, МП-2УМ.
Горелка газовая.
Ступка агатовая.
Палочка платиновая или алюминиевая.
Секундомер, ГОСТ 5072-79.
Щипцы тигельные с платиновым наконечником.
Воронки из полиэтилена.
Пробирки колориметрические, ГОСТ 10515-75.
Колбы мерные, 4 — 25, ГОСТ 1770-74Е, вместимостью 25, 100 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 10 и 50 мл.
Сосуды полиэтиленовые.
Реактивы, растворы, материалы
Кварц чистый (горный хрусталь).
Натрий двууглекислый, ГОСТ 4201-79, хч.
Натрий хлористый, ГОСТ 4233-77, хч.
Кислота соляная, ГОСТ 3118-77, хч, разбавленная 1:1.
Кислота винная, ГОСТ 5817-77, чда, 10% раствор, свежеприготовленный.
Кислота азотная, ГОСТ 4461-77, хч, разбавленная 1:1.
Кислота аскорбиновая, ГОСТ 4815-76, хч, 1% раствор, свежеприготовленный.
Аммоний хлористый, ГОСТ 3773-72, хч, 2% раствор.
Аммоний молибденовокислый, ГОСТ 3765-78, хч, перекристаллизованный, 10% раствор. Соль растворяется в горячей воде. (В случае выпадения осадка раствор фильтруют.)
Составной плавень готовят из свежевысушенных при 105 град. C равных весовых частей двууглекислого и хлористого натрия, тщательно растертых и перемешанных в агатовой ступке. Плавень хранят в склянке с притертой пробкой.
Стандартный раствор N 1 с содержанием диоксида кремния 200 мкг/мл. Навеску тонко измельченного в агатовой ступке кварца 0,0200 г помещают в платиновый тигель и тщательно перемешивают с 1,0 г составного плавня. Тигель устанавливают в муфельную печь и сплавляют при температуре 900 град. C в течение 4 минут (по секундомеру), плав выщелачивают в несколько приемов водой в мерную колбу вместимостью 100 мл и доводят объем водой до метки. Раствор хранят в полиэтиленовом сосуде в течение одного месяца.
Стандартный раствор N 2, содержащий 20 мкг/мл диоксида кремния, готовят соответствующим разбавлением стандартного раствора N 1 водой. Применяют свежеприготовленный раствор.
Все реактивы хранят в полиэтиленовой посуде.
Фильтры АФА-ХП или АФА-ВП, ТУ 95-743-80.
Фильтры беззольные бумажные «синяя лента», диаметром 4 см, ТУ 6-09-1678-77.
Отбор пробы воздуха
Воздух с объемным расходом 15 л/мин. аспирируют через фильтр АФА. Для измерения 1/2 ПДК следует отобрать 100 л воздуха. Пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 5 часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ДИОКСИДА КРЕМНИЯ
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание диоксида кремния, мкг |
| 1 | 0 | 10,0 | 0 |
| 2 | 0,1 | 9,9 | 2,0 |
| 3 | 0,2 | 9,8 | 4,0 |
| 4 | 0,3 | 9,7 | 6,0 |
| 5 | 0,5 | 9,5 | 10,0 |
| 6 | 0,7 | 9,3 | 14,0 |
| 7 | 0,9 | 9,1 | 18,0 |
| 8 | 1,2 | 8,8 | 24,0 |
| 9 | 1,5 | 8,5 | 30,0 |
| 10 | 2,0 | 8,0 | 40,0 |
| 11 | 2,5 | 7,5 | 50,0 |
Затем во все пробирки шкалы добавляют по 1,0 мл раствора молибдата аммония и перемешивают. Через 5 мин. добавляют по 1,0 мл раствора винной кислоты, спустя 10 минут по 0,25 мл раствора аскорбиновой кислоты, перемешивают после добавления каждого реактива. Через 20 минут измеряют оптическую плотность растворов при длине волны 600 нм по отношению к контролю (раствор N 1 по таблице). Измерения проводят в кюветах с толщиной поглощающего слоя 1 см.
Строят градуировочный график зависимости оптической плотности от содержания диоксида кремния в градуировочных растворах (мкг).
Проверку градуировочного графика проводят 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
Для отделения цветных и тяжелых металлов, приводящих к коррозии платиновых тиглей, фильтр с пробой обрабатывают раствором кислот. Для этого фильтр заливают в химическом стакане вместимостью 50 мл 10 мл смеси из равных объемов разбавленных 1:1 соляной и азотной кислот и кипятят в течение 2 — 3 минут. Раствор фильтруют через небольшой фильтр «синяя лента» и отбрасывают. Стакан промывают дважды 10 мл воды, перенося фильтр АФА и смывы на фильтр «синяя лента».
Бумажный фильтр вместе с фильтром АФА помещают в платиновый тигель, который устанавливают в холодную муфельную печь, и осторожно высушивают, обугливают и озоляют при 500 — 600 град. C.
По охлаждении в тигель вносят 0,5 г составного плавня, который тщательно перемешивают с золой, после чего тигель закрывают крышкой и помещают в нагретую до 900 град. C печь, сплавляют до получения однородного плава в течение 10 минут. Плав выщелачивают в несколько приемов водой, раствор плава переносят в мерную колбу вместимостью 25 мл и доводят объем до метки водой. Если раствор мутный, его фильтруют.
Для анализа отбирают в колориметрическую пробирку 1,0 мл раствора, который обрабатывают и фотометрируют аналогично градуировочным растворам.
Предварительно проводят холостой опыт для проверки степени чистоты тиглей, фильтров и плавня. Значение оптической плотности холостого опыта не должно превышать 0,020.
Содержание диоксида кремния в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию диоксида кремния в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентраций фтористого водорода и солей фтористоводородной кислоты
Характеристика метода
Определение основано на взаимодействии фтор-иона с комплексом циркония и ксиленолового оранжевого, сопровождающемся ослаблением окраски.
Отбор проб проводится с концентрированием на фильтр типа АФА (для улавливания фторидов) и последовательно присоединенную к фильтродержателю сорбционную трубку (для улавливания фтористого водорода).
Нижний предел измерения содержания фтор-иона в объеме анализируемого раствора 0,5 мкг.
Нижний предел измерения фтористого водорода в воздухе 0,1 мг/куб. м, хорошо растворимых фторидов 0,25 мг/куб. м, плохо растворимых фторидов 1,0 мг/куб. м (при отборе 10 л воздуха).
Диапазон измеряемых концентраций для фтористого водорода от 0,1 до 5,0 мг/куб. м для хорошо растворимых фторидов от 0,25 до 12,5 мг/куб. м, для плохо растворимых фторидов от 1,0 до 20 мг/куб. м.
Определению не мешают марганец, железо, хром, никель, медь, кальций в количестве до 0,4 мг, алюминий — до 0,15 мг в анализируемом объеме.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения для фтористого водорода — 1 — 1,5 часа, для растворимых фторидов — 2 часа, для нерастворимых фторидов — 5 часов, включая отбор пробы 5 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр ФЭК-56М, ГОСТ 15150-74, 1-й класс, или другой марки по ГОСТ 12083-78.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Тигли платиновые, ГОСТ 6563-75.
Щипцы тигельные с платиновыми наконечниками.
Печь муфельная, МП-2УМ.
Ступка агатовая.
Баня водяная.
Груша резиновая.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 и 1000 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-80Е, вместимостью 50 мл.
Колбы конические, ГОСТ 25336-80Е, вместимостью 50 и 100 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 и 50 мл.
Воронки стеклянные, ГОСТ 25336-82Е.
Бутыли полиэтиленовые, ТУ 6-19-45-74, вместимостью 100, 250, 500 мл.
Сорбционная трубка СТ 212, ТУ 25-1110.039.20-84. При отсутствии трубок заводского производства их изготавливают следующим образом. Стеклянную трубку длиной 150 — 170 мм и диаметром 8 — 10 мм из стекла «Пирекс» запаивают с одного конца и формируют в днище 5 — 6 отверстий диаметром 1 мм. В трубку засыпают 2 мл заранее подготовленного носителя — стеклянных гранул размером 1 — 2 мм и формируют вторую перфорированную перегородку, фиксирующую слой носителя. Для получения стеклянных гранул стеклодрот из стекла «Пирекс» дробят на шаровой мельнице или в ступке. Дробление в ступке проводят под слоем воды. Высушенные гранулы просеивают через сита, например почвенные, и собирают фракцию с размером гранул 1 — 2 мм. Гранулы тщательно промывают соляной кислотой (1:2) при кипячении, водопроводной и дистиллированной водой и высушивают.
Готовые поглотительные трубки моют раствором соляной кислоты при кипячении, затем водой до отрицательной реакции на хлор-ионы и высушивают в сушильном шкафу. Промывание удобно проводить с помощью водоструйного насоса.
Полиэтиленовая Г-образная трубка, диаметром 7 — 10 мм, длиной 7 — 10 см.
Реактивы, растворы, материалы
Все реактивы готовят на бидистиллированной воде.
Натрия фторид, ГОСТ 4463-76, хч, перекристаллизованный и высушенный до постоянного веса.
Соляная кислота, ГОСТ 3118-77, хч, концентрированная, 1 н. и 3,5 н. (точно) растворы. Титр 3,5 н. раствора устанавливают по титру 3,5 н. едкого натра в присутствии фенолфталеина.
Натрия гидроксид, ГОСТ 4328-77, хч, 3,5 н. раствор. Концентрацию едкого натра устанавливают путем титрования 1 н. соляной кислотой, приготовленной из фиксанала, в присутствии фенолфталеина.
Этиловый спирт, ректификат, ТУ 6-09-1710-77, хч, 60% раствор.
Фенолфталеин, ГОСТ 5850-72, чда, 0,1% раствор в 60% растворе этилового спирта.
Цирконил азотнокислый, двуводный, ТУ 6-09-1406-76, чда, раствор, содержащий 0,08 мг/мл цирконила. Навеску 0,080 г соли растворяют в 3,5 н. соляной кислоте в мерной колбе вместимостью 1 л. Раствор устойчив в течение 6 месяцев.
Ксиленоловый оранжевый, ТУ 6-09-1509-78, чда, 0,2% и 0,02% растворы. Для приготовления 0,2% раствора 100 мг индикатора растворяют в 50 мл воды. Раствор готов к употреблению через сутки. Устойчив при хранении в холодильнике не более 2-х недель.
0,02% раствор ксиленолового оранжевого готовят в день анализа разбавлением водой 0,2% раствора в 10 раз.
Натрий углекислый безводный, ГОСТ 83-79, хч.
Диоксид кремния, ГОСТ 9428-73, чда, мелкоистолченный в агатовой ступке порошок.
Раствор плава для приготовления градуировочных растворов для определения нерастворимых фторидов. 2,5 углекислого натрия и 0,1 г диоксида кремния помещают в платиновый тигель, закрывают крышкой и сплавляют в муфельной печи при температуре 900 град. C до получения однородного плава. По охлаждении плав растворяют в горячей бидистиллированной воде и переводят раствор в мерную колбу вместимостью 250 мл. Доводят объем до метки и перемешивают.
Глицерин, ГОСТ 6259-75, чда.
Калий углекислый безводный, ГОСТ 422-75, хч.
Поглотительный раствор для обработки сорбционных трубок: в мерную колбу вместимостью 50 мл вносят 1,5 г углекислого калия, 7,5 мл глицерина и доводят раствор до метки бидистиллированной водой.
Стандартный раствор N 1, содержащий 100 мкг/мл фтор-иона, готовят растворением 0,2210 г фторида натрия в бидистиллированной воде в мерной колбе вместимостью 1 л. Раствор устойчив в течение 2 недель при хранении в полиэтиленовой бутыли.
Стандартный раствор N 2, содержащий 5 мкг/мл фтора, готовят соответствующим разбавлением стандартного раствора N 1 водой, свежеприготовленный.
Фильтры АФА-ХП-20 или АФА-ВП-20, ТУ 95.743-80.
Фильтры бумажные беззольные «синяя лента», ТУ 6-09-1678-77.
Отбор пробы воздуха
Подготовка сорбционных трубок к отбору проб.
Перед отбором проб слой носителя в сорбционной трубке обрабатывают поглотительным раствором. Для этого с помощью пипетки вносят в трубку 0,2 мл раствора и равномерно распределяют его по поверхности стеклянных гранул с помощью резиновой груши, надетой на выходной конец трубки. Подготовленные к отбору проб трубки закрывают заглушками, маркируют с помощью отрезков лейкопластыря с номером, наклеенных на расстоянии 1,5 — 2,0 см от выходного отверстия трубок, и помещают в полиэтиленовый пакет.
Воздух с объемным расходом 2 л/мин. аспирируют через фильтр АФА и последовательно соединенные с фильтродержателем с помощью полиэтиленовой Г-образной трубки 2 сорбционные трубки, обработанные раствором углекислого калия и глицерина. Во время отбора пробы сорбционные трубки устанавливают вертикально.
Для измерения 1/2 ПДК фтористого водорода следует отобрать 10 л воздуха. Пробы сохраняются в течение 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблицам 1 и 2.
Шкала градуировочных растворов для определения фтористого водорода готовится аналогично таблице 1, но при этом в каждую пробирку шкалы добавляют 0,2 мл поглотительного раствора.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ХОРОШО РАСТВОРИМЫХ ФТОРИДОВ
| N стандарта | Стандартный раствор N 2, мл | Бидистиллированная вода, мл | Содержание фтор-иона в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,1 | 4,9 | 0,5 |
| 3 | 0,2 | 4,8 | 1,0 |
| 4 | 0,4 | 4,6 | 2,0 |
| 5 | 0,8 | 4,2 | 4,0 |
| 6 | 1,2 | 3,8 | 6,0 |
| 7 | 1,6 | 3,4 | 8,0 |
| 8 | 2,0 | 3,0 | 10,0 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ПЛОХО РАСТВОРИМЫХ ФТОРИДОВ
| N стандарта | Стандартный раствор N 2, мл | Бидистиллированная вода, мл | Содержание фтор-иона в градуировочном растворе, мкг |
| 1 | 0 | 2,5 | 0 |
| 2 | 0,1 | 2,4 | 0,5 |
| 3 | 0,2 | 2,3 | 1,0 |
| 4 | 0,4 | 2,1 | 2,0 |
| 5 | 0,8 | 1,7 | 4,0 |
| 6 | 1,2 | 1,3 | 6,0 |
| 7 | 1,6 | 0,9 | 8,0 |
| 8 | 2,0 | 0,5 | 10,0 |
Во все пробирки шкалы для определения плохо растворимых фторидов приливают 2,5 мл раствора плава и осторожно перемешивают.
Растворы всех трех шкал обрабатывают одинаково: прибавляют по 1,6 мл азотнокислого цирконила, через 5 минут по 1,6 мл 0,02-процентного раствора ксиленолового оранжевого, перемешивают и через 3 — 5 минут измеряют оптическую плотность растворов при длине волны 540 нм в кюветах с толщиной слоя 2 см по отношению к соответствующему контрольному раствору (растворы N 1 по таблицам). Готовят не менее 3-х контрольных растворов для каждой шкалы.
За 3 — 5 минут до измерения оптической плотности контрольные растворы, относящиеся к определенной градуировочной шкале, сливают в одну пробирку и перемешивают.
В связи с тем, что с увеличением содержания фтор-ионов в градуировочных растворах наблюдается ослабление окраски, для получения градуировочной кривой, исходящей из нулевой точки, необходимо изменить порядок измерения оптической плотности. Для этого контрольный раствор помещают в левый световой пучок фотоэлектроколориметра, а анализируемый раствор — в правый.
Строят градуировочные графики зависимости оптической плотности градуировочных растворов от содержания фтор-иона для определения фтористого водорода и фторидов с использованием соответствующих шкал.
Проверка градуировочных графиков проводится 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Определение фтористого водорода
Содержимое сорбционных трубок анализируют отдельно, результаты суммируют.
Сорбционную трубку тщательно вытирают фильтровальной бумагой и помещают в пробирку слоем стеклянных гранул вниз. Вводят внутрь трубки 10 мл бидистиллированной воды и через 10 — 15 мин. путем 5 — 8-кратного прокачивания ее через слой стеклянных гранул с помощью резиновой груши (не допуская попадания раствора внутрь груши), переводят пробу в раствор. Затем трубку удаляют из пробирки, вытесняя остатки раствора грушей, отбирают на анализ 2 — 5 мл раствора в колориметрические пробирки, доводят объем раствора до 5 мл водой и обрабатывают и фотометрируют аналогично градуировочным растворам.
Определение растворимых фторидов
Фильтр с отобранной пробой помещают в стакан вместимостью 50 мл, наливают около 8 мл горячей бидистиллированной воды, осторожно перемешивают и фильтруют через беззольный бумажный фильтр в мерную колбу вместимостью 25 мл. Операцию повторяют трижды. После охлаждения объем в колбе доводят до 25 мл водой. На анализ отбирают в колориметрические пробирки 2 — 5 мл раствора пробы, доводят объем до 5 мл водой и далее обрабатывают и анализируют аналогично градуировочным растворам.
Определение нерастворимых фторидов
Фильтр АФА после удаления с него растворимых фторидов и беззольный фильтр, через который проводилось фильтрование, помещают в платиновый тигель, осторожно обугливают и озоляют в муфельной печи при температуре 500 град. C. Затем тигель охлаждают, добавляют в него 500 мг углекислого натрия и 10 мг двуокиси кремния и выдерживают в муфельной печи при температуре 900 град. C в течение часа. После сплавления пробу растворяют в горячей воде, постепенно переводя ее в мерную колбу вместимостью 50 мл. Для анализа в колориметрические пробирки отбирают аликвотную часть 2,5 мл, доводят объем до 5 мл водой, прибавляют 1,6 мл раствора азотнокислого цирконила и тщательно перемешивают. Пробы выдерживают 30 минут на кипящей водяной бане для устранения влияния алюминия. После охлаждения добавляют 1,6 мл 0,02-процентного раствора ксиленолового оранжевого и через 3 — 5 минут фотометрируют аналогично градуировочным растворам.
Содержание фтористого водорода и фторидов в анализируемом растворе пробы (мкг) определяют по соответствующему градуировочному графику.
Концентрацию фтористого водорода и фторидов в воздухе (по фтор-иону) рассчитывают по формуле 1 (Приложение 4.2).
Измерение концентрации озона
Метод 1
Характеристика метода
Метод основан на реакции озонолиза эвгенола с образованием формальдегида, поглощении последнего раствором серной кислоты с последующим определением по реакции с фенилгидразином.
Отбор проб проводится на систему, включающую сорбционную трубку с пленкой раствора эвгенола в уксусной кислоте и последовательно соединенные с ней поглотительные приборы, заполненные раствором серной кислоты.
Нижний предел измерения содержания озона в объеме анализируемого раствора 0,57 мкг.
Нижний предел измерения озона в воздухе 0,04 мг/куб. м (при отборе 30 л воздуха).
Диапазон измеряемых концентраций от 0,04 до 2,0 мг/куб. м.
Определению озона не мешает двуокись азота. Определению мешает формальдегид.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 50 минут, включая отбор пробы 10 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр ФЭК-56М, ГОСТ 15150-74, 1-й класс, или другой марки по ГОСТ 12083-78.
Аспирационное устройство.
Сорбционная трубка СТ 212, ТУ 25-1110.039.20-84. (Трубка из стекла «Пирекс» диаметром 10 мм, длиной 200 мм со слоем стеклянной крошки (2 мл) размером 1 — 2 мм, помещенным между двумя перфорированными перегородками.)
Поглотительные приборы Рыхтера, длиной 170 мм, диаметром 20 мм.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25 и 50 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Пробирки химические, ГОСТ 25336-82Е, размером 15 x 100 мм.
Пробирки колориметрические, ГОСТ 10515-75, размером 15 x 100 мм.
Колбы конические, ГОСТ 25336-82, вместимостью 200 мл.
Г-образная стеклянная трубка, диаметром 0,6 — 1,0 см и длиной колен 2,5 — 3 см.
Реактивы, растворы, материалы
Формалин, СТУ 43-182-61, 40% раствор.
Йод, ТУ 6-09-2540-72, 0,1 н. раствор, готовят из фиксанала.
Натрий серноватистокислый (тиосульфат натрия), ТУ 6-09-2540-72, 0,1 н. раствор, готовят из фиксанала.
Натрия гидроксид, ГОСТ 4328-77, хч, 10% раствор.
Кислота соляная, ГОСТ 3118-77, хч, 10% раствор.
Крахмал растворимый, ГОСТ 10163-62, ч, 0,5-процентный раствор.
Кислота уксусная, ледяная, ГОСТ 61-75, хч.
4-аллил-2-метоксифенол (эвгенол), ТУ 18-16-134-83, 10% раствор в уксусной кислоте.
Кислота серная, ГОСТ 4204-77, хч, 20% раствор (по весу) и 0,02 М раствор.
Этиловый спирт, ГОСТ 5962-67, ректификат.
Фенилгидразин солянокислый, ГОСТ 5834-73, чда, 1% раствор, свежеприготовленный, отфильтрованный.
Хлорамин Б, ТУ 6-09-3021-73, хч, 0,5% раствор, отфильтрованный. Устойчив в течение недели.
Основной раствор формальдегида. Готовят 1% раствор формалина и количество формальдегида в нем определяют титриметрически. В колбу 200 мл вводят 1 мл 1-процентного раствора формалина, приливают 10 мл воды и добавляют из бюретки 10 мл раствора йода. Затем по каплям прибавляют 10% раствор гидроксида натрия до получения светло-желтого окрашивания. Оставляют на 10 мин., затем добавляют 2 мл 10% раствора соляной кислоты до полного выделения йода, опять оставляют на 10 мин. и оттитровывают раствор тиосульфатом натрия с крахмалом в качестве индикатора. В таких же условиях оттитровывают и контрольный раствор. Разность между объемом раствора тиосульфата, пошедшим на контрольное титрование и титрование раствора формалина, равно количеству мл раствора йода, пошедшего на окисление формальдегида. 1 мл 0,1 н. раствора йода соответствует 1,5 мг формальдегида. Рассчитывают содержание формальдегида в 1 мл основного раствора.
Стандартный раствор формальдегида N 1 с концентрацией 1 мг/мл готовят соответствующим разбавлением основного раствора формальдегида 0,02 М раствором серной кислоты. Раствор устойчив в течение не менее 6 месяцев.
Стандартные растворы формальдегида N 2 с концентрацией 10 мкг/мл и N 3 с концентрацией 1 мкг/мл готовят соответствующим разбавлением раствора N 1 0,02 М раствором серной кислоты непосредственно перед употреблением.
Отбор пробы воздуха
Подготовка сорбционных трубок к отбору проб воздуха.
Трубки промывают с помощью водоструйного насоса или резиновой груши растворами обычных моющих средств, затем водопроводной и дистиллированной водой, после чего сушат при температуре 120 — 150 град. C.
Слой стекла в вертикально поставленной входным отверстием вверх сорбционной трубке смачивают 0,15 мл уксуснокислого раствора эвгенола. С помощью груши, надетой на выходной конец трубки, равномерно распределяют раствор по поверхности стеклянной крошки. Обработанная сорбционная трубка сохраняется в течение суток.
Подготовленная сорбционная трубка используется для однократного отбора пробы воздуха.
Воздух с объемным расходом 3 л/мин. аспирируют через сорбционную трубку, установленную вертикально входным отверстием вниз, соединенную посредством Г-образной трубки с двумя последовательно соединенными поглотительными приборами, содержащими по 5 мл 0,02 М раствора серной кислоты.
Пробы хранятся в течение суток. Для измерения 1/2 ПДК озона следует отобрать 30 л воздуха.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 20 минут) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОЗОНА
| N стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Серная кислота, 0,02 М раствор, мл | Содержание формальдегида в градуировочном растворе, мкг |
| 1 | 0 | — | 2,5 | 0 |
| 2 | 0,2 | — | 2,3 | 0,2 |
| 3 | 0,5 | — | 2,0 | 0,5 |
| 4 | 1,0 | — | 1,5 | 1,0 |
| 5 | 2,0 | — | 0,5 | 2,0 |
| 6 | — | 0,3 | 2,2 | 3,0 |
| 7 | — | 0,5 | 2,0 | 5,0 |
| 8 | — | 0,7 | 1,8 | 7,0 |
| 9 | — | 1,0 | 1,5 | 10,0 |
Во все пробирки шкалы прибавляют по 0,5 мл этилового спирта и по 0,5 мл раствора солянокислого фенилгидразина. Содержимое пробирок осторожно перемешивают и спустя 15 минут добавляют по 0,5 мл раствора хлорамина. Выдерживают 10 минут, прибавляют по 1,0 мл серной кислоты, перемешивают и через 10 минут измеряют оптическую плотность растворов при длине волны 400 нм в кюветах с толщиной поглощающего слоя 10 мм относительно контрольного раствора. Соблюдение сроков внесения реактивов и фотометрирования обязательно.
Строят градуировочный график зависимости оптической плотности растворов от содержания в них формальдегида (мкг).
Проверка градуировочного графика проводится 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Содержимое поглотительных приборов анализируют отдельно, результаты суммируют. Из каждого поглотительного прибора в колориметрические пробирки вносят по 2,5 мл раствора пробы, доводят объем до 5,0 мл 0,02 М раствором серной кислоты и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание формальдегида в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию озона в воздухе (C мг/куб. м) вычисляют по формуле:
![]() ,
,
где:
а — содержание формальдегида в анализируемом объеме раствора пробы, найденное по градуировочному графику, мкг;
в — общий объем раствора пробы, мл;
б — объем раствора пробы, взятый для анализа, мл;
![]() — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л;
— объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л;
1,6 — коэффициент пересчета формальдегида на озон;
1,77 — эмпирический коэффициент, характеризующий выход формальдегида в результате реакции озона с эвгенолом.
Метод 2
Характеристика метода
Метод основан на реакции озона с йодистым калием с выделением йода, который образует окрашенный продукт с солянокислым диметил-n-фенилендиамином.
Отбор проб проводится с концентрированием в раствор йодида калия.
Нижний предел измерения содержания в объеме анализируемого раствора 0,4 мкг.
Нижний предел измерения в воздухе 0,05 мг/куб. м (при отборе 10 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,05 мг/куб. м до 1,3 мг/куб. м.
Влияние окислов азота устраняется в процессе отбора пробы.
Суммарная погрешность измерения не превышает +/- 15%.
Время выполнения измерения 40 — 45 минут, включая отбор пробы 20 минут.
Приборы, аппаратура, посуда
Фотоколориметр ФЭК-56, ГОСТ 15150-74, 1-й класс, или другой марки по ГОСТ 12083-78.
Аспирационное устройство.
Печь муфельная, МП-2УМ.
Поглотительные приборы Рыхтера, длиной 170 мм, диаметром 20 мм.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 25336-84Е, вместимостью 1, 2, 5 и 10 мл.
Цилиндры мерные, ГОСТ 1770-74Е.
Колбы мерные, ГОСТ 1774-74Е, вместимостью 100 и 1000 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 и 500 мл.
Воронка стеклянная, ГОСТ 25336-82Е.
Воронка Бюхнера, ГОСТ 25336-82Е.
Термометр на 100 град. C.
Эксикатор, ГОСТ 25336-82.
Фильтрующий патрон: стеклянная трубка диаметром 6 мм, длиной 80 мм, имеющая дно с отверстиями 0,5 мм. Со стороны перфорированного дна в трубку помещают слой силикагеля длиной 5 мм, далее слой поглотительного порошка длиной 60 мм и слой полихрома длиной 2 — 3 мм, слегка уплотненный.
Г-образная стеклянная трубка, диаметром 6 мм и длиной колен 2,5 — 3 см.
Реактивы, растворы, материалы
Йод, 0,01 н. раствор, готовят из фиксанала, ТУ 6-09-2540-72.
Калий йодистый, ГОСТ 4232-74, хч, 1% раствор, хранят в склянке из темного стекла.
Спирт этиловый, ректификованный, ГОСТ 18300-72.
N,N-Диметил-n-фенилендиамин дигидрохлорид, ТУ 6-09-1903-77, чда, 0,02% раствор в этаноле. Готовят в перекристаллизованной соли. Раствор устойчив в течение 2 недель. Для перекристаллизации диметил-n-фенилендиамина солянокислого 6 г вещества вносят в термостойкий стакан, добавляют 90 мл 96% спирта и помещают в водяную баню, имеющую температуру 90 — 98 град. C.
Содержимое стакана перемешивают стеклянной палочкой до полного растворения вещества. Раствор быстро фильтруют через складчатый фильтр, фильтрат помещают на водяную баню с температурой 90 град. C. После закипания раствора быстро и осторожно добавляют ацетон до заметного помутнения раствора (160 мл). Раствор снова помещают на водяную баню, перемешивают и потирают стеклянной палочкой по стенкам стакана до выпадания осадка.
Затем к раствору с осадком добавляют 120 мл ацетона и помещают стакан в емкость со льдом. Через 10 — 15 минут раствор декантируют с осадка. К осадку добавляют 60 мл ацетона, перемешивают и осадок отфильтровывают через бумажный фильтр на воронке Бюхнера. Осадок с фильтра переносят в стакан, прибавляют 60 мл спирта и повторяют операцию перекристаллизации, как описано выше.
Осадок после второй перекристаллизации промывают на воронке Бюхнера под вакуумом ацетоном дважды по 50 мл. Полученный продукт сушат между листами фильтровальной бумаги, помещают в эксикатор над прокаленной окисью кальция и защищают от света. Высушенную соль хранят в склянке с притертой пробкой в темноте.
Натрия гидроксид, ГОСТ 4328-77, хч.
Азотная кислота, ГОСТ 4461-77, хч, 0,1 н. и 10% растворы.
Серебро азотнокислое, ГОСТ 1277-75, хч, 1% раствор.
Ацетон, ГОСТ 2603-71, чда.
Соляная кислота, ГОСТ 3118-77, хч, 25% и 0,1 н. растворы.
Серная кислота, ГОСТ 4204-77, хч.
Хрома (VI) оксид (хромовый ангидрид), ГОСТ 3776-78, чда.
Кальций хлористый плавленный, ГОСТ 4460-77.
Силикагель марки АСК, фракция 0,25 — 0,5 мм; силикагель кипятят в течение часа с 25% раствором соляной кислоты, затем отмывают водой до отрицательной реакции на ион хлора (реакция с раствором азотнокислого серебра). Силикагель сушат в сушильном шкафу, а затем прокаливают в муфельной печи при 480 — 500 град. C в течение 2-х часов. Хранят в банке с притертой пробкой.
Поглотительный порошок для отделения мешающих примесей.
К 47 г силикагеля, нагретого до 35 — 40 град. C, добавляют при непрерывном перемешивании 20 мл раствора хромового ангидрида в концентрированной серной кислоте (на 20 мл кислоты — 0,6 г хромового ангидрида) до получения однородной массы оранжевого цвета; полученный поглотительный порошок хранят в банке с притертой пробкой в эксикаторе над прокаленным хлоридом кальция.
Стандартный раствор йода с концентрацией 10 мкг/мл готовят перед употреблением в мерной колбе вместимостью 100 мл; вносят 0,8 мл 0,01 н. раствора йода и доводят до метки 1% раствором йодистого калия.
Фильтры бумажные беззольные «синяя лента», ТУ 6-09-1678-77.
Отбор пробы воздуха
Воздух с объемным расходом 0,5 л/мин. аспирируют через фильтрующий патрон и последовательно соединенные с ним 2 поглотительных прибора, содержащих по 7 мл 1% раствора йодистого калия.
Фильтрующий патрон должен располагаться вертикально, дном с отверстиями вниз.
Для измерения 1/2 ПДК озона следует отобрать 10 л воздуха.
Пробы не хранятся.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 15 минут) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОЗОНА
| N стандарта | Стандартный раствор йода, мл | Раствор йодида калия, 1%, мл | Содержание в градуировочном растворе, мкг | |
| йода | озона | |||
| 1 | 0 | 5,0 | 0 | 0 |
| 2 | 0,2 | 4,8 | 2,0 | 0,378 |
| 3 | 0,4 | 4,6 | 4,0 | 0,756 |
| 4 | 0,6 | 4,4 | 6,0 | 1,134 |
| 5 | 0,8 | 4,2 | 8,0 | 1,512 |
| 6 | 1,0 | 4,0 | 10,0 | 1,89 |
Во все пробирки шкалы прибавляют по 0,5 мл 0,02% раствора солянокислого диметилпарафенилендиамина, перемешивают, выдерживают 10 мин. и измеряют оптическую плотность растворов при длине волны 490 нм в кюветах с толщиной слоя 1 см относительно контрольного раствора (раствор N 1 по таблице).
Строят градуировочный график зависимости оптической плотности раствора от содержания озона (мкг).
Проверка градуировочного графика проводится 1 раз в квартал или в случае использования новой партии реактивов.
Проведение измерения
1 — 5 мл раствора из поглотительного прибора вносят в пробирку, доводят объем до 5,0 мл 1% раствором йодида калия и далее обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание озона в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику.
Концентрацию озона в воздухе рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета йода на озон — 0,189.
Измерение концентрации оксидов азота (II) и (IV)
Характеристика метода
Метод основан на реакции взаимодействия оксидов азота (VI) с реактивом Грисса-Илосвая с образованием окрашенного в красный цвет соединения.
Для определения оксида азота (II) его окисляют в ходе отбора пробы до оксида азота (IV) раствором, содержащим перманганат калия и серную кислоту.
Отбор проб проводится с концентрированием в раствор йодида калия.
Нижний предел измерения содержания оксида азота (IV) в анализируемом объеме пробы 0,3 мкг, оксида (II) — 0,2 мкг.
Нижний предел измерения оксида азота (IV) в воздухе 1 мг/куб. м, оксида азота (II) — 0,65 мг/куб. м (при отборе 0,6 л воздуха).
Диапазон измеряемых концентраций оксида азота (IV) — от 1 до 42 мг/куб. м, оксида азота (II) — от 0,65 до 27 мг/куб. м.
Измерению не мешают другие газовые компоненты сварочного аэрозоля.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения анализа, включая время отбора пробы, 40 мин.
Приборы, аппаратура, посуда
Фотоэлектроколориметр ФЭК-56М, ГОСТ 15150-74, 1-й класс, или другой марки по ГОСТ 12083-78.
Аспирационное устройство.
Поглотительные приборы с пористой пластинкой.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 100 и 250 мл.
Реактивы, растворы, материалы
Натрий азотнокислый, ГОСТ 4197-74, хч.
Калий йодистый, ГОСТ 42-32-74, хч, 8% раствор (хранится в темной склянке).
Натрий сернистокислый, ГОСТ 195-77, 0,01 н. раствор свежеприготовленный.
Кислота сульфаниловая, ГОСТ 5821-78, чда.
Кислота уксусная, ГОСТ 61-75, хч, 10% раствор.
Альфа-нафтиламин, ГОСТ 8827-74, чда.
Реактив Грисса-Илосвая:
а) 0,83 г сульфаниловой кислоты растворяют в мерной колбе, вместимостью 250 мл, в 10-процентной уксусной кислоте;
б) 0,166 г — альфа-нафтиламина помещают в колбу и добавляют 30 мл дистиллированной воды, колбу нагревают на кипящей водяной бане до образования на дне лиловой капли. Раствор фильтруют в мерную колбу вместимостью 250 мл и доводят объем до метки 10% уксусной кислотой.
Растворы «а» и «б» сохраняют в склянках темного стекла с притертыми пробками. Перед употреблением для получения реактива Грисса-Илосвая оба раствора смешивают в соотношении 1:1.
Кислота серная, ГОСТ 4204-77, разбавленная 1:2.
Калий марганцевокислый, ГОСТ 20490-75, хч, 5% раствор.
Окислительный раствор: к 20 мл раствора марганцевокислого калия приливают 2 мл серной кислоты; этого количества окислительной смеси достаточно для отбора 10 проб воздуха. Реактив готовится в день отбора проб.
Стандартный раствор оксида азота (IV) N 1 с концентрацией 100 мкг/мл готовят растворением навески 0,0150 г азотистокислого натрия в 8% растворе йодида калия в мерной колбе вместимостью 100 мл. Раствор устойчив в течение месяца.
Стандартный раствор оксида азота N 2 с концентрацией 1 мкг/мл готовят разбавлением раствора N 1 в 100 раз 8% раствором йодида калия. Применяют свежеприготовленным.
Фильтры бумажные обеззоленные, ТУ 6-09-1678-77.
Отбор пробы воздуха
Воздух со скоростью 0,1 л/мин. аспирируют через систему, состоящую из 3-х поглотительных приборов — 1 и 3, содержащих по 10 мл 8% раствора йодида калия, и поглотителя с 20 мл окислительной смеси (2), помещенного между ними. В первом поглотителе идет поглощение оксида азота (IV) из воздуха, во втором — проходит окисление оксидов азота (II) из воздуха до оксида азота (IV), который поглощается в 3-м поглотителе. Для измерения 1/2 ПДК оксида азота (IV) следует отобрать 0,6 л воздуха. Пробы не хранятся.
Подготовка к измерению
Градуировочный раствор (устойчив в течение 5 часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОКСИДА АЗОТА
| N градуировочного раствора | Стандартный раствор N 2, мл | Йодид калия, 8% раствор, мл | Содержание оксида азота (IV) в градуировочном растворе, мкг |
| 1 | 0 | 5,0 | 0 |
| 2 | 0,3 | 4,7 | 0,3 |
| 3 | 1,0 | 4,0 | 1,0 |
| 4 | 2,0 | 3,0 | 2,0 |
| 5 | 4,0 | 1,0 | 4,0 |
| 6 | 5,0 | 0,0 | 5,0 |
В пробирки шкалы добавляют по 1 мл реактива Грисса-Илосвая. Через 15 — 20 мин. во все пробы приливают по 0,1 мл раствора сульфита натрия, перемешивают и измеряют оптическую плотность окрашенных растворов при длине волны 510 — 540 нм в кювете с толщиной слоя 1 см относительно контрольного раствора (раствор N 1 таблицы).
Строят градуировочный график зависимости оптической плотности растворов от содержания в них оксида азота (IV) (мкг), проверку которого проводят 1 раз в квартал или при использовании новой партии реактивов.
Проведение измерения
Раствор пробы из первого и третьего поглотителя, в количестве 2 и 5 мл, вносят в колориметрические пробирки; 2 мл пробы доводят до объема 5 мл раствором йодида калия. Далее растворы обрабатывают и фотометрируют аналогично градуировочным растворам.
Содержание оксида азота (IV) в анализируемом растворе пробы определяют по градуировочному графику.
Концентрацию оксида азота (IV) в воздухе (мг/куб. м) рассчитывают по формуле 1 (Приложение 4.2).
Коэффициент пересчета содержания оксида азота (IV) в оксид азота (II) — 0,65.
3.2. ПОЛЯРОГРАФИЧЕСКИЕ МЕТОДЫ
Измерение концентраций меди, никеля, кадмия, цинка
Характеристика метода
Метод основан на восстановлении ионов меди, никеля, кадмия и цинка на ртутно-капельном электроде на фоне хлоридно-аммиачного буферного раствора в присутствии сульфита натрия.
Отбор проб воздуха проводится с концентрированием на фильтр АФА.
Потенциал восстановления относительно донной ртути меди — 0,35 В, никеля — 0,92 В, кадмия — 0,66 В, цинка — 1,2 В.
Нижний предел измерения в полярографируемом растворе составляет для меди, никеля, кадмия и цинка 0,1 мкг/мл.
Нижний предел измерения указанных металлов в воздухе 0,02 мг/куб. м (при отборе 150 л воздуха).
Диапазон измеряемых концентраций в воздухе для меди от 0,02 до 7,0 мг/куб. м, никеля от 0,02 до 8,0 мг/куб. м, кадмия от 0,02 до 3,0 мг/куб. м и цинка от 0,02 до 7,0 мг/куб. м.
Определению не мешает алюминий, свинец, серебро, кремний. Железо не мешает в количестве до 20 мкг в полярографируемом объеме пробы.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 4 часа, включая отбор пробы воздуха 10 минут.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутно-капельным электродом (с записью полярограмм в переменно-токовом и постоянно-токовом режимах), ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня водяная.
Тигли фарфоровые, ГОСТ 9147-80Е.
Чашки фарфоровые, ГОСТ 9147-80Е.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 и 1000 мл.
Пипетки мерные, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Реактивы, растворы, материалы
Медь металлическая, электролитическая, ГОСТ 859-78.
Никель металлический, ГОСТ 849-70.
Кадмий гранулированный, ТУ 6-09-3097-78, чда.
Цинк металлический, гранулированный, ГОСТ 989-75, хч.
Аммиак водный, ГОСТ 3760-79, хч, 25% раствор.
Аммония хлорид, ГОСТ 3773-72, хч.
Натрий сернистокислый, ГОСТ 195-77, чда, насыщенный раствор, свежеприготовленный.
Кислота соляная, ГОСТ 3118-77, хч, концентрированная.
Кислота азотная, ГОСТ 4461-77, хч, концентрированная, 1:1 и 3:2.
Стандартный раствор меди N 1 с концентрацией 1 мг/мл меди. 1,0000 г электролитической меди помещают в фарфоровую чашку и растворяют при нагревании в 20 — 25 мл азотной кислоты (1:1). Раствор выпаривают до небольшого объема (2 — 3 мл), приливают 15 мл концентрированной соляной кислоты и вновь выпаривают до небольшого объема. Выпаривание проводят 2 раза, каждый раз с 5 мл соляной кислоты. Растворенный в воде остаток количественно переносят в мерную колбу вместимостью 1000 мл. К раствору добавляют 50 мл соляной кислоты и доводят до метки дистиллированной водой. Раствор устойчив более года.
Стандартный раствор меди N 2 с концентрацией 10 мкг/мл готовят разбавлением раствора N 1 дистиллированной водой и применяют свежеприготовленным.
Стандартный раствор никеля N 1 с концентрацией 1 мг/мл: 1,000 г металлического никеля растворяют при нагревании на водяной бане в фарфоровой чашке в 10 мл разбавленной азотной кислоты (3:2). Содержимое чашки упаривают до объема 3 мл, растворяют в 30 — 40 мл воды и количественно переносят в мерную колбу вместимостью 1000 мл. Доводят объем раствора до метки дистиллированной водой и перемешивают. Раствор устойчив более года.
Стандартный раствор никеля N 2 с концентрацией 10 мкг/мл готовят соответствующим разбавлением раствора N 1 дистиллированной водой и применяют свежеприготовленным.
Стандартный раствор кадмия N 1 с концентрацией 1 мг/мл. Навеску 1,0000 г кадмия помещают в фарфоровую чашку и растворяют при нагревании в 20 — 25 мл азотной кислоты (1:1). Раствор упаривают до небольшого объема (2 — 3 мл), приливают 15 мл концентрированной соляной кислоты и вновь упаривают до небольшого объема. Выпаривание повторяют еще 2 раза каждый раз с 5 мл соляной кислоты. После охлаждения в чашку приливают 50 мл соляной кислоты, количественно переносят раствор в мерную колбу вместимостью 1000 мл, доводят раствор до метки водой и тщательно перемешивают. Раствор устойчив более года.
Стандартный раствор кадмия N 2 с концентрацией 10 мкг/мл готовят разбавлением стандартного раствора N 1 дистиллированной водой и применяют свежеприготовленным.
Стандартный раствор цинка N 1 с концентрацией 1 мг/мл получают растворением 1,0000 г металлического гранулированного цинка в 15 мл концентрированной соляной кислоты. Раствор переносят в мерную колбу вместимостью 10000 мл и доводят до метки водой. Раствор устойчив более года.
Стандартный раствор цинка N 2 с концентрацией 10 мкг/мл готовят разбавлением стандартного раствора N 1 дистиллированной водой и применяют свежеприготовленным.
Хлоридно-аммиачный буферный раствор: 50 г хлорида аммония растворяют в дистиллированной воде в мерной колбе вместимостью 1000 мл, добавляют 25 г сернистокислого натрия, 75 мл 25% раствора аммиака и объем доводят до метки водой. Раствор устойчив в течение 2 недель.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр АФА-ХА-20, ТУ 95.743-80.
Бумага индикаторная универсальная, ТУ 6-09-1181-76.
Отбор проб воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА-ХА-20, укрепленный в фильтродержателе. Для измерения 1/2 ПДК меди, никеля, кадмия и цинка достаточно отобрать 150 л воздуха. Пробы хранятся в течение 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2 часов) готовят согласно таблицам 1 — 4.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МЕДИ
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание меди в градуировочном растворе, мкг |
| 1 | 0 | 8,5 | 0 |
| 2 | 0,1 | 8,4 | 1,0 |
| 3 | 0,2 | 8,3 | 2,0 |
| 4 | 0,5 | 8,0 | 5,0 |
| 5 | 1,0 | 7,5 | 10,0 |
| 6 | 2,0 | 6,5 | 20,0 |
| 7 | 3,0 | 5,5 | 30,0 |
| 8 | 4,0 | 4,5 | 40,0 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ НИКЕЛЯ
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание никеля в градуировочном растворе, мкг |
| 1 | 0 | 8,5 | 0 |
| 2 | 0,1 | 8,4 | 1,0 |
| 3 | 0,2 | 8,3 | 2,0 |
| 4 | 0,5 | 8,0 | 5,0 |
| 5 | 1,0 | 7,5 | 10,0 |
| 6 | 2,0 | 6,5 | 20,0 |
| 7 | 3,0 | 5,5 | 30,0 |
| 8 | 4,0 | 4,5 | 40,0 |
| 9 | 5,0 | 3,5 | 50,0 |
Таблица 3
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ КАДМИЯ
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание кадмия в градуировочном растворе, мкг |
| 1 | 0 | 8,5 | 0 |
| 2 | 0,1 | 8,4 | 1,0 |
| 3 | 0,2 | 8,3 | 2,0 |
| 4 | 0,5 | 8,0 | 5,0 |
| 5 | 1,0 | 7,5 | 10,0 |
| 6 | 1,5 | 7,0 | 15,0 |
| 7 | 2,0 | 6,5 | 20,0 |
Таблица 4
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЦИНКА
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание цинка в градуировочном растворе, мкг |
| 1 | 0 | 8,5 | 0 |
| 2 | 0,1 | 8,4 | 1,0 |
| 3 | 0,2 | 8,3 | 2,0 |
| 4 | 0,5 | 8,0 | 5,0 |
| 5 | 1,0 | 7,5 | 10,0 |
| 6 | 2,0 | 6,5 | 20,0 |
| 7 | 3,0 | 5,5 | 30,0 |
| 8 | 4,0 | 4,5 | 40,0 |
Растворы градуировочных шкал оттитровываются по каплям раствором аммиака до pH 8 (по индикаторной бумаге). Затем в пробирки добавляют по 1 мл хлоридно-аммиачного буфера, по 0,5 мл свежеприготовленного насыщенного раствора сульфита натрия, доводят объем до 10 мл водой, перемешивают, выдерживают 10 минут, переносят в электролитическую ячейку и полярографируют.
Режим полярографирования переменно-токовый (на примере ПУ-1), поляризующее напряжение от -0,15 до -1,5 В; амплитуда квадратно-волновой формы 30 мВ; задержка 4,1 с; скорость развертки 5 мВ/с; поляризация катодная; диапазон тока 0,5 x 1; координаты самописца: X = 2 x 100 мВ/см, Y = 1 x 100 мкА/см.
Высоту пика меди измеряют при E1/2 = -0,35 В; никеля при E1/2 = -0,92 В; кадмия при E1/2 = -0,66 В; цинка при E1/2 = -1,2 В.
Строят градуировочные графики зависимости высот пиков меди, никеля, кадмия и цинка (мм) от содержания этих веществ в растворе (мкг).
Проверка градуировочного графика проводится 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой осторожно переносят в фарфоровый тигель и озоляют в муфельной печи при температуре 500 — 600 град. C в течение 30 минут. После охлаждения золу растворяют в 5 мл концентрированной соляной кислоты. Содержимое тигля выпаривают до влажных солей на кипящей водяной бане, остаток растворяют в нескольких миллилитрах горячей воды и переносят количественно в мерную колбу вместимостью 25 мл, обмывая тигель водой. Раствор в колбе доводят до метки водой и перемешивают.
1 — 8 мл полученного раствора вносят в пробирку, доводят объем до 8,5 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам.
Содержание меди, кадмия, никеля и цинка в анализируемом объеме раствора пробы (мкг) определяют по соответствующим градуировочным графикам или методом добавок.
Концентрацию металлов в воздухе (мг/куб. м) рассчитывают по формулам 1 или 4, 5 (Приложение 4.2 или 4.3).
Измерение концентраций меди, никеля и кобальта
Характеристика метода
Метод основан на восстановлении диметилглиоксиматных комплексов меди, никеля, кобальта на ртутно-капельном электроде на фоне хлоридно-аммиачного буферного раствора в присутствии сульфита натрия. Потенциал восстановления относительно донной ртути меди — 0,25 В; никеля — 0,86 В; кобальта — 1,02 В.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения в полярографируемом растворе составляет для меди 0,3 мкг/мл, кобальта 0,02 мкг/мл, никеля 0,01 мкг/мл.
Нижний предел измерения в воздухе меди 0,5 мг/куб. м, кобальта 0,03 мг/куб. м, никеля 0,02 мг/куб. м (при отборе 50 л воздуха).
Диапазон измеряемых концентраций в воздухе для меди от 0,5 до 10 мг/куб. м, для никеля от 0,02 до 1,0 мг/куб. м, для кобальта от 0,03 до 1,0 мг/куб. м.
Измерению не мешают алюминий, кремний, свинец, цинк.
Железо не мешает определению при содержании более 20 мкг в полярографируемом объеме раствора.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерений 4 часа, включая отбор проб 4 минуты.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутно-капельным электродом с записью полярограмм в переменно-токовом режиме, ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня водяная.
Чашки фарфоровые, ГОСТ 9147-80Е.
Тигли фарфоровые, ГОСТ 9147-80Е.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100, 500 и 1000 мл.
Пипетки мерные, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Реактивы, растворы, материалы
Медь металлическая, электролитическая, ГОСТ 859-78.
Кобальт сернокислый, семиводный, ГОСТ 4462-78, хч.
Никель металлический, ГОСТ 849-70, марка Н.О.
Аммиак водный, ГОСТ 3760-79, хч, 25% и 1 М растворы.
Аммоний хлористый, ГОСТ 3773-72, хч.
Натрий сернистокислый, ГОСТ 195-77, хч, безводный.
Кислота азотная, ГОСТ 4461-77, хч.
Кислота соляная, ГОСТ 3118-77, хч, концентрированная.
Хлоридно-аммиачный буферный раствор: 50 г хлористого аммония растворяют в дистиллированной воде в мерной колбе вместимостью 1000 мл, добавляют 25 г сернистокислого натрия, 75 мл 25% раствора аммиака и объем раствора доводят до метки.
Диметилглиоксим, ГОСТ 5828-77, хч.
4 x ![]() М раствор диметилглиоксима в 1 М аммиаке готовят растворением 0,018 г диметилглиоксима в 40 мл 1 М аммиака.
М раствор диметилглиоксима в 1 М аммиаке готовят растворением 0,018 г диметилглиоксима в 40 мл 1 М аммиака.
Стандартный раствор меди N 1. 1,0000 г металлической меди помещают в фарфоровую чашку и растворяют при нагревании в 20 — 25 мл азотной кислоты (1:1). Раствор выпаривают до небольшого объема (2 — 3 мл), приливают 15 мл концентрированной соляной кислоты и вновь выпаривают до небольшого объема. Выпаривание проводят 2 раза, каждый раз с 5 мл соляной кислоты, переливают раствор в мерную колбу вместимостью 100 мл и доводят объем до метки дистиллированной водой. В 1 мл полученного раствора содержится 1 мг меди. Раствор устойчив более года.
Стандартные растворы меди N 2 с концентрацией 10 мкг/мл готовят путем соответствующего разбавления водой стандартного раствора N 1 и применяют свежеприготовленным.
Стандартный раствор никеля N 1. 1,0000 г металлического никеля растворяют при нагревании на водяной бане в фарфоровой чашке в 10 мл разбавленной азотной кислоты (3:2). Содержимое чашки выпаривают до объема 3 мл, растворяют в 30 — 40 мл воды и доводят объем раствора до метки в мерной колбе вместимостью 1000 мл. В 1 мл раствора содержится 1 мг никеля. Раствор устойчив более года.
Стандартные растворы никеля N 2 и N 3 с концентрациями соответственно 10 мкг/мл и 1 мкг/мл готовят путем соответствующего разбавления водой стандартных растворов N 1 и N 2 и применяют свежеприготовленными.
Стандартный раствор кобальта N 1 с концентрацией 1 мг/мл. 4,7700 г сернокислого кобальта растворяют в мерной колбе вместимостью 1000 мл в дистиллированной воде.
Стандартные растворы кобальта N 2 и N 3 с концентрациями соответственно 10 мкг/мл и 1 мкг/мл готовят путем соответствующего разбавления водой стандартных растворов N 1 и применяют свежеприготовленными.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр типа АФА, ТУ 95.743-80.
Бумага индикаторная универсальная, ТУ 6-09-1181-76.
Отбор пробы воздуха
Воздух с объемным расходом 15 л/мин. аспирируют через фильтр АФА.
Для измерения 1/2 ПДК меди, никеля, кобальта следует отобрать 50 л воздуха. Пробы хранятся не менее 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 45 минут) готовят согласно таблицам 1 — 3.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МЕДИ
| N стандарта | Стандартный раствор N 2, мл | Вода дистиллированная, мл | Содержание меди в градуировочном растворе, мкг |
| 1 | 0 | 8,2 | 0 |
| 2 | 0,3 | 7,9 | 3,0 |
| 3 | 0,4 | 7,8 | 4,0 |
| 4 | 0,5 | 7,7 | 5,0 |
| 5 | 0,6 | 7,6 | 6,0 |
| 6 | 0,7 | 7,5 | 7,0 |
| 7 | 0,8 | 7,4 | 8,0 |
| 8 | 0,9 | 7,3 | 9,0 |
| 9 | 1,0 | 7,2 | 10,0 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ НИКЕЛЯ
| N стандарта | Стандартный раствор N 3, мл | Вода дистиллированная, мл | Содержание никеля в градуировочном растворе, мкг |
| 1 | 0 | 8,2 | 0 |
| 2 | 0,1 | 8,1 | 0,1 |
| 3 | 0,2 | 8,0 | 0,2 |
| 4 | 0,3 | 7,9 | 0,3 |
| 5 | 0,4 | 7,8 | 0,4 |
| 6 | 0,5 | 7,7 | 0,5 |
| 7 | 0,6 | 7,6 | 0,6 |
| 8 | 0,7 | 7,5 | 0,7 |
| 9 | 0,8 | 7,4 | 0,8 |
| 10 | 0,9 | 7,3 | 0,9 |
| 11 | 1,0 | 7,2 | 1,0 |
Таблица 3
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ КОБАЛЬТА
| N стандарта | Стандартный раствор N 3, мл | Вода дистиллированная, мл | Содержание кобальта в градуировочном растворе, мкг |
| 1 | 0 | 8,2 | 0 |
| 2 | 0,2 | 8,0 | 0,2 |
| 3 | 0,3 | 7,9 | 0,3 |
| 4 | 0,4 | 7,8 | 0,4 |
| 5 | 0,5 | 7,7 | 0,5 |
| 6 | 0,6 | 7,6 | 0,6 |
| 7 | 0,7 | 7,5 | 0,7 |
| 8 | 0,8 | 7,4 | 0,8 |
| 9 | 0,9 | 7,3 | 0,9 |
| 10 | 1,0 | 7,2 | 1,0 |
Во все пробирки шкал для определения меди, никеля и кобальта добавляют по 0,3 мл раствора диметилглиоксима, по 1 мл хлоридно-аммиачного буфера, по 0,5 мл свежеприготовленного насыщенного раствора сернистокислого натрия и перемешивают. Перед проведением измерений градуировочные растворы выдерживают не менее 15 минут.
Градуировочные растворы заливают в электролизер и полярографируют. Режим полярографирования переменно-токовый (на примере ПУ-1), поляризующее напряжение от -0,1 В до -1,5 В; амплитуда квадратно-волновой формы 30 мВ; задержка — 3,8 с; скорость развертки 5 мВ/с; поляризация катодная: диапазон тока — 0,5 x 1; координаты самописца: X = 2 x 100 мВ/см; Y = 5 x 10 мкА/см.
Высоту пика меди измеряют при потенциале E1/2 = -0,25 В, никеля — 0,86 В, кобальта — 1,02 В.
Строят градуировочные графики зависимости высот пиков меди, никеля и кобальта (мм) от их содержания в растворе (мкг).
Проверка градуировочного графика проводится 1 раз в неделю или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в фарфоровый тигель и озоляют в муфельной печи при температуре 500 град. C в течение 30 минут.
После охлаждения зольный остаток растворяют в 5 мл концентрированной соляной кислоты, упаривают до влажных солей, переносят в мерную колбу вместимостью 25 мл, доводят объем до метки водой и перемешивают.
Отбирают в пробирку 0,5 — 3 мл раствора пробы, доводят объем до 10 мл дистиллированной водой и оттитровывают раствор концентрированным раствором аммиака до pH 9 (по индикаторной бумаге) (1 — 3 капли).
Далее раствор обрабатывают и полярографируют аналогично градуировочным растворам.
Содержание меди, никеля и кобальта в анализируемом объеме раствора пробы (мкг) определяют по соответствующему градуировочному графику или методом добавок.
Концентрацию вещества в воздухе (мг/куб. м) рассчитывают по формулам 1 или 4, 5 (Приложение 4.2 или 4.3).
Измерение концентрации железа, титана, молибдена, оксидов хрома (III и VI) и ванадия
Характеристика метода
Определение основано на переведении металлов и их оксидов в раствор (при этом хром (III) в процессе окислительного охлаждения пробы переходит в хром (VI) с последующим полярографированием раствора на ртутно-капельном электроде в переменно-токовом режиме). В качестве фонового электролита для определения железа, молибдена, титана и хрома (VI) применяют ацетатный буферный раствор с pH 5,5 в присутствии Трилона Б. Потенциал восстановления железа — 0,23 В, титана — 0,6 В, молибдена — 0,9 В, хрома (VI) — 1,25 В относительно донной ртути.
Определение ванадия проводят на фоне аммиачно-ацетатного буферного раствора с pH 9,5 в присутствии Трилона Б. Потенциал восстановления ванадия — 1,23 В.
На фоновом электролите с pH 5,5 хром и ванадий выходят одним пиком при потенциале — 1,25 В, а при pH 9,5 определение ванадия селективно в присутствии хрома.
С целью раздельного определения того и другого элемента пользуются тремя градуировочными графиками, два из которых (на хром и ванадий) строят на фоновом электролите с pH 5,5 и одним (на ванадий) — при pH 9,5. Определение хрома проводится по разности двух измерений.
Отбор проб воздуха проводится с концентрированием на фильтр АФА.
Нижний предел измерения ионов металлов в полярографируемом растворе составляет для железа 1 мкг/мл, титана 0,1 мкг/мл, молибдена 0,2 мкг/мл, хрома (VI) 0,1 мкг/мл, ванадия 0,1 мкг/мл.
Нижний предел измерения в воздухе железа 2 мг/куб. м, титана 0,2 мг/куб. м, молибдена 0,4 мг/куб. м, хрома (VI) 0,005 мг/куб. м, хрома (III) 0,2 мг/куб. м, ванадия 0,06 мг/куб. м (при отборе 300 л воздуха).
Диапазон измеряемых концентраций для железа от 2 до 83 мг/куб. м, титана от 0,2 до 104 мг/куб. м, молибдена от 0,4 до 104 мг/куб. м, хрома (VI) от 0,005 до 0,1 мг/куб. м, хрома (III) от 0,2 до 42 мг/куб. м, ванадия от 0,04 до 8,3 мг/куб. м.
Определению не мешают другие сопутствующие металлы.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 5 часов, включая время отбора пробы 15 минут.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутно-капельным электродом (с записью полярограмм в переменно-токовом и постоянно-токовом режимах), ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня водяная.
Баня песчаная.
Секундомер, ГОСТ 5072-79.
Тигли фарфоровые, ГОСТ 9147-80Е.
Чашки фарфоровые, ГОСТ 9147-80Е.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 и 1000 мл.
Пробирки колориметрические с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 мл.
Секундомер, ГОСТ 5072-79.
Реактивы, растворы, материалы
Железо металлическое восстановленное, ТУ 6-09-2227-72, хч.
Титана двуокись, ТУ 6-09-2166-77, чда.
Аммоний молибденовокислый, 4-водный, ГОСТ 3765-78, хч.
Калий двухромовокислый, ГОСТ 4220-75, хч.
Аммоний метаванадиевокислый (ванадат аммония), ГОСТ 9336-75, чда.
Калий сернокислый пиро (пиросульфат калия), ГОСТ 7172-76, чда.
Кислота соляная, ГОСТ 3118-77, хч.
Натрий углекислый, безводный, ГОСТ 83-79, хч.
Калий азотнокислый, ГОСТ 4217-77, хч.
Кислота серная, ГОСТ 4204-77, хч, 5% и 10% растворы.
Кислота уксусная ледяная, ГОСТ 61-75, хч, 4 М раствор.
Аммиак водный, ГОСТ 3760-79, хч, 25% раствор.
Натрий уксуснокислый, 3-водный, ГОСТ 199-78, хч.
Трилон Б, ГОСТ 10682-73, хч, 0,1 М раствор.
Ацетатный буферный раствор (pH 5,5) готовят следующим образом: растворяют в воде 49 г ацетата натрия, добавляют 20 мл 4 М уксусной кислоты и объем доводят до 200 мл дистиллированной водой.
Плавень (смесь солей для получения плава). Готовят растиранием в ступке двух частей карбоната натрия и одной части нитрата калия. Хранят в склянке с притертой пробкой.
Стандартный раствор железа N 1 с концентрацией 0,2 мг/мл готовят растворением 0,2000 г металлического железа в 10 мл концентрированной соляной кислоты при нагревании. Раствор количественно переносят в мерную колбу вместимостью 1000 мл и доводят до метки дистиллированной водой. Устойчив в течение 6 месяцев.
Стандартный раствор железа N 2 с концентрацией 10 мкг/мл готовят соответствующим разбавлением стандартного раствора N 1 дистиллированной водой. Используют свежеприготовленный раствор.
Стандартный раствор титана N 1 с концентрацией 100 мкг/мл готовят сплавлением 0,1668 г диоксида титана, высушенного до постоянного веса, на газовой горелке с 6,0 г пиросульфата калия и растворением плава в 5% серной кислоте в мерной колбе вместимостью 1000 мл. Устойчив в течение 6 месяцев.
Стандартный раствор титана N 2 с концентрацией 10 мкг/мл готовят разбавлением раствора N 1 в 10 раз дистиллированной водой непосредственно перед употреблением.
Стандартный раствор молибдена N 1 с концентрацией 1 мг/мл готовят растворением 0,1845 г молибдата аммония в воде в мерной колбе вместимостью 100 мл. Устойчив в течение 6 месяцев.
Стандартный раствор молибдена N 2 с концентрацией 10 мкг/мл готовят соответствующим разбавлением стандартного раствора N 1 в 100 раз дистиллированной водой непосредственно перед употреблением.
Стандартный раствор хрома (VI) N 1 с концентрацией, соответствующей концентрации оксида хрома (VI) 1 мг/мл, готовят растворением 0,1471 г дважды перекристаллизованного бихромата калия в воде в мерной колбе вместимостью 1000 мл. Устойчив в течение 6 месяцев.
Стандартный раствор хрома (VI) N 2 с концентрацией, соответствующей концентрации оксида хрома (VI) 10 мкг/мл, готовят разбавлением стандартного раствора N 1 в 100 раз дистиллированной водой непосредственно перед употреблением.
Стандартный раствор ванадия N 1 с концентрацией, соответствующей концентрации оксида ванадия (V) 1 мг/мл, готовят растворением 1,2860 г метаванадата аммония в воде, содержащей 0,2 мл аммиака, в мерной колбе вместимостью 1000 мл. Раствор устойчив в течение 6 месяцев.
Стандартный раствор ванадия N 2 с концентрацией, соответствующей концентрации оксида ванадия (V) 10 мкг/мл, готовят разбавлением раствора N 1 в 100 раз дистиллированной водой в день анализа.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Индикаторная бумага универсальная, ТУ 6-09-1181-76.
Фильтры обеззоленные «синяя лента», ТУ 6-09-1678-77, диаметром 4 см.
Фильтры АФА-ХА-20, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 20 л/мин. аспирируют через фильтр АФА-ХА-20, помещенный в фильтродержатель. Для определения 1/2 ПДК следует отобрать 300 л воздуха. Пробы не следует хранить из-за возможных потерь шестивалентного хрома.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблицам 1 — 6.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЖЕЛЕЗА
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Концентрация железа в градуировочном растворе, мкг/мл |
| 1 | 0 | 7,0 | 0 |
| 2 | 1,0 | 6,0 | 1,0 |
| 3 | 2,0 | 5,0 | 2,0 |
| 4 | 3,0 | 4,0 | 3,0 |
| 5 | 4,0 | 3,0 | 4,0 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ТИТАНА
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Концентрация титана в градуировочном растворе, мкг/мл |
| 1 | 0 | 7,0 | 0 |
| 2 | 0,1 | 6,9 | 0,1 |
| 3 | 0,2 | 6,8 | 0,2 |
| 4 | 0,5 | 6,5 | 0,5 |
| 5 | 1,0 | 6,0 | 1,0 |
| 6 | 2,0 | 5,0 | 2,0 |
| 7 | 3,0 | 4,0 | 3,0 |
| 8 | 4,0 | 3,0 | 4,0 |
| 9 | 5,0 | 2,0 | 5,0 |
Таблица 3
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МОЛИБДЕНА
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Концентрация молибдена в градуировочном растворе, мкг/мл |
| 1 | 0 | 7,0 | 0 |
| 2 | 0,2 | 6,8 | 0,2 |
| 3 | 0,5 | 6,5 | 0,5 |
| 4 | 1,0 | 6,0 | 1,0 |
| 5 | 2,0 | 5,0 | 2,0 |
| 6 | 3,0 | 4,0 | 3,0 |
| 7 | 4,0 | 3,0 | 4,0 |
| 8 | 5,0 | 2,0 | 5,0 |
Таблица 4 — 5
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОКСИДОВ ХРОМА ИЛИ ВАНАДИЯ
| N стандарта | Стандартный раствор N 2, хрома или ванадия, мл | Дистиллированная вода, мл | Концентрация оксида хрома (VI) или оксида ванадия (V) в градуировочном растворе, мкг/мл |
| 1 | 0 | 7,0 | 0 |
| 2 | 0,1 | 6,9 | 0,1 |
| 3 | 0,2 | 6,8 | 0,2 |
| 4 | 0,5 | 6,5 | 0,5 |
| 5 | 1,0 | 6,0 | 1,0 |
| 6 | 1,5 | 5,5 | 1,5 |
| 7 | 2,0 | 5,0 | 2,0 |
Таблица 6
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОКСИДА ВАНАДИЯ (V) ПРИ pH 9,5
| N стандарта | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Концентрация оксида ванадия (V) в градуировочном растворе, мкг/мл |
| 1 | 0,0 | 7,0 | 0 |
| 2 | 0,1 | 6,9 | 0,1 |
| 3 | 0,2 | 6,8 | 0,2 |
| 4 | 0,5 | 6,5 | 0,5 |
| 3 | 1,0 | 6,0 | 1,0 |
| 6 | 1,5 | 5,5 | 1,5 |
| 7 | 2,0 | 5,0 | 2,0 |
Во все пробирки шкал N 1 — 5 добавляют по 2 мл ацетатного буферного раствора, 1 мл Трилона Б и перемешивают. Градуировочные растворы заливают в электролизер, продувают 5 минут аргоном и полярографируют в диапазоне потенциалов -0,1 + -1,5 В. Режим полярографирования (на примере ПУ-1) 2-электродный ТАСТ-режим, амплитуда квадратно-волновой формы 30 мВ, задержка 3,6 с, скорость развертки 5 мВ/с, поляризация катодная, диапазон тока 0,5 x 1. Координаты самописца X = 2 x 100 мВ/см, Y = 5 x 10 мкА/см.
Во все пробирки шкалы N 6 (на ванадий) добавляют по 2 мл ацетатного буфера, 1 мл Трилона Б, 0,15 мл аммиака и перемешивают (pH полученного раствора ~ 9,5).
Градуировочные растворы заливают в электролизер, продувают аргоном в течение 5 мин. и полярографируют в диапазоне потенциалов -0,9 + -1,5 В в тех же условиях, что и для остальных градуировочных шкал.
Строят градуировочные графики зависимости высот пиков железа, молибдена, титана, оксида хрома (VI) и оксида ванадия (V) (мм) от концентрации в растворе (мкг/мл) при pH 5,5 и график зависимости высоты пика ванадия от его концентрации в растворе (мкг/мл) при pH 9,5.
Проверку градуировочных графиков проводят 1 раз в месяц или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в стаканчик, отмывают несколькими порциями горячей (40 — 50 град. C) воды, фильтруют через небольшой беззольный фильтр и по охлаждении доводят объем раствора до 10 мл водой. Фильтрат используют для определения водорастворимого хрома (VI). Для этого отбирают 6 мл фильтрата, доводят объем до 7,0 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам шкалы N 6.
Фильтр с нерастворившейся частью пробы и бумажный фильтр переносят в фарфоровый тигель, осторожно высушивают и озоляют при температуре 550 град. C. Зольный остаток смешивают с 0,5 г плавня и помещают тигель в охлажденную до 300 град. C муфельную печь, постепенно повышая температуру до 850 — 860 град. C. Сплавляют в течение 30 минут до получения однородного плава. Плав растворяют в 10% серной кислоте, дважды упаривая до влажных солей. Содержимое тигля растворяют в небольшом количестве 10% серной кислоты и количественно переносят в мерную колбу вместимостью 50 мл. Объем раствора в колбе доводят до метки 10% серной кислотой (раствор А).
10 мл раствора А переносят в мерную колбу вместимостью 25 мл, нейтрализуют раствором аммиака до pH 5,0 (по индикаторной бумаге), доводят до метки водой и перемешивают (раствор Б).
Для определения железа, титана, молибдена и хрома (III) отбирают в пробирку 0,2 — 2 мл раствора Б, доводят до 7,0 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам шкал 1 — 5.
Для определения ванадия 1 — 6 мл раствора Б вносят в пробирку, нейтрализуют раствор аммиаком по каплям до pH 7, доводят объем до 7,0 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам шкалы N 6 (pH конечного раствора должен быть 9,5).
Концентрацию оксида хрома (VI) в анализируемом объеме раствора пробы (мкг/мл) определяют по градуировочному графику N 4.
Концентрацию железа, титана, молибдена и оксида ванадия (V) (мкг/мл) определяют соответственно по градуировочным графикам N 1, 2, 3 и 6.
Для расчета концентрации оксида хрома (III) по градуировочному графику N 6 (при pH 9,5) определяют концентрацию оксида ванадия (V) в анализируемом растворе (мкг/мл). Находят соответствующую этой концентрации высоту пика оксида ванадия (V) по градуировочному графику N 5 для определения оксида ванадия (V) при pH 5,5 (h1), которую вычитают из величины пика оксида хрома (VI) и оксида ванадия (V), полученного при полярографировании раствора пробы с pH 5,5 (h). Значение разности высот пиков (h — h1) соответствует концентрации оксида хрома (VI) в полярографируемом растворе пробы, которую определяют по градуировочному графику N 4.
Для определения концентрации оксида хрома (III) полученное значение умножают на коэффициент 0,76.
Концентрацию металлов и их оксидов в воздухе (мг/куб. м) рассчитывают по формуле 3 (Приложение 4.2).
Измерение концентрации железа и марганца
Характеристика метода
Метод основан на восстановлении ионов железа и марганца на ртутно-капельном электроде на фоне раствора триэтаноламина с добавлением соляной кислоты и едкого натра.
Отбор проб воздуха проводится с концентрированием на фильтр АФА.
Потенциал восстановления относительно донной ртути железа — 1,06 В; марганца — 0,5 В.
Нижний предел измерения в полярографируемом растворе составляет для железа 0,2 мкг/мл, для марганца 0,2 мкг/мл.
Нижний предел измерения в воздухе железа 0,05 мг/куб. м, марганца 0,05 мг/куб. м (при отборе 200 л воздуха).
Диапазон измеряемых концентраций для железа и марганца от 0,05 до 12,5 мг/куб. м.
Определению железа мешает хром.
Определению марганца мешает медь.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 3,5 часа, включая отбор пробы 15 мин.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутным капельным электродом с записью полярограмм в переменно-токовом режиме, ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня песчаная.
Ступка фарфоровая, ГОСТ 9147-80Е.
Тигли фарфоровые, ГОСТ 9147-80Е, с крышками.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100, 250, 500 и 1000 мл.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Воронки стеклянные, ГОСТ 25336-82Е.
Палочки стеклянные, ГОСТ 25336-82Е.
Пипетки, ГОСТ 20292-74Е, вместимостью 0,2, 1, 2, 5 и 10 мл.
Реактивы, растворы, материалы
Железо металлическое восстановленное, ТУ 6-09-2227-72, хч.
Марганца сульфат, 5-водный, ТУ 6-09-01-218-74, чда.
Триэтаноламин, ТУ 6-09-2448-72, ч, 0,5 М раствор.
Натрия гидроксид, ГОСТ 4328-77, хч, 10% раствор.
Кислота соляная, ГОСТ 3118-77, концентрированная и 0,5 М раствор.
Кислота серная, ГОСТ 4204-77, чда, 10% раствор.
Кислота азотная, ГОСТ 4461-77, хч, концентрированная.
Аммиак водный, ГОСТ 3760-79, чда, 25% раствор.
Натрия карбонат, безводный, ГОСТ 83-79, хч.
Калия нитрат, ГОСТ 4217-77, хч.
Плавень (смесь солей для получения плавня) готовят растиранием в фарфоровой ступке 2 частей карбоната натрия и 1 части нитрата калия. Плавень хранят в банке с притертой пробкой.
Стандартный раствор железа N 1: навеску металлического железа 0,2000 г растворяют в 10 мл концентрированной соляной кислоты при нагревании на песчаной бане, количественно переносят в мерную колбу вместимостью 1000 мл и доводят дистиллированной водой до метки. В 1 мл раствора содержится 200 мкг железа. Раствор устойчив в течение месяца.
Стандартный раствор железа N 2 с концентрацией 10 мкг/мл готовят разбавлением в 20 раз раствора N 1 водой, свежеприготовленный.
Стандартный раствор марганца N 1: 4,3880 г сульфата марганца растворяют в воде в мерной колбе вместимостью 1000 мл и доводят дистиллированной водой до метки. В 1 мл раствора содержится 1 мг марганца. Устойчив в течение 6 месяцев.
Стандартный раствор марганца N 2 с концентрацией 10 мкг/мл готовят разбавлением в 100 раз раствора N 1 водой, свежеприготовленный.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр АФА-ХП-20, ТУ 95.743-80.
Бумага индикаторная лакмусовая красная, ТУ 6-09-3403-78.
Отбор пробы воздуха
Воздух с объемным расходом 15 л/мин. аспирируют через укрепленный в фильтродержателе фильтр АФА-ХП-20.
Для измерения 1/2 ПДК железа и марганца следует отобрать 200 л воздуха. Отобранные пробы хранятся в течение 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х час.) готовят согласно таблицам 1 — 2.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ЖЕЛЕЗА
| N стандарта | Стандартный раствор железа N 2, мл | Дистиллированная вода, мл | Содержание железа в градуировочном растворе, мкг |
| 1 | 0 | 7,5 | 0 |
| 2 | 0,2 | 7,3 | 2,0 |
| 3 | 0,4 | 7,1 | 4,0 |
| 4 | 0,6 | 6,9 | 6,0 |
| 5 | 0,8 | 6,7 | 8,0 |
| 6 | 1,0 | 6,5 | 10,0 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МАРГАНЦА
| N стандарта | Стандартный раствор марганца N 2, мл | Дистиллированная вода, мл | Содержание марганца в градуировочном растворе, мкг |
| 1 | 0 | 7,5 | 0 |
| 2 | 0,2 | 7,3 | 2,0 |
| 3 | 0,4 | 7,1 | 4,0 |
| 4 | 0,6 | 6,9 | 6,0 |
| 5 | 0,8 | 6,7 | 8,0 |
| 6 | 1,0 | 6,5 | 10,0 |
Во все пробирки шкал добавляют по 1 мл 0,5 М соляной кислоты, по 1 мл 0,5 М триэтаноламина, по 0,5 мл 10% гидроксида натрия и перемешивают содержимое. Градуировочные растворы переносят в электролизер, продувают аргоном в течение 5 мин. и полярографируют.
Режим полярографирования переменно-токовый (на примере ПУ-1), трехэлектродный ТАСТ-режим: поляризующее напряжение от -0,3 В до -1,3 В; амплитуда квадратно-волновой формы 12 мВ; задержка 3,8 с; скорость развертки 9 мВ; поляризация катодная; диапазон тока 0,5 x 1; координаты самописца: X = 2 x 100 мВ/см; Y = 1 x 100 мкА/см.
Высоту пика железа измеряют при потенциале E1/2 = -1,06 В, марганца — 0,5 В.
Строят градуировочные графики зависимости высот пиков железа и марганца (мм) от их содержания в анализируемом растворе (мкг).
Проверка градуировочных графиков проводится 1 раз в неделю или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с пробой помещают в фарфоровый тигель, накрывают крышкой и осторожно озоляют. Зольный остаток по охлаждении тщательно перемешивают и растирают лопаточкой с 0,5 г плавня. Далее тигель помещают в охлажденную до 350 — 400 град. C муфельную печь, повышают температуру до 800 — 850 град. C, при которой выдерживают тигель в течение 25 — 30 минут до полного сплавления смеси.
После охлаждения тигля содержимое растворяют в 25 мл 10% серной кислоты. 0,1 — 5 мл пробы доводят до 7,5 мл водой и нейтрализуют аммиаком (по лакмусу).
Далее обработка пробы и полярографирование проводятся аналогично градуировочным растворам.
Содержание железа и марганца в анализируемом объеме раствора пробы (мкг) определяют по соответствующему градуировочному графику или методом добавок.
Концентрацию железа и марганца в воздухе (в мг/куб. м) рассчитывают по формулам 1, 4 и 5 (Приложения 4.2, 4.3).
Коэффициент пересчета железа на оксид железа — 1,43.
Измерение концентрации вольфрама
Метод основан на восстановлении вольфрамат-ионов на ртутно-капельном электроде на фоне раствора, содержащего хлорат калия, серную и миндальную кислоты.
Потенциал восстановления вольфрамат-ионов относительно насыщенного каломельного электрода — 0,6 В.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения в полярографируемом растворе составляет 0,05 мкг/мл.
Нижний предел измерения в воздухе 2,5 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций для вольфрама от 2,5 до 40 мг/куб. м.
Определению вольфрама не мешает присутствие 100-кратных по отношению к вольфраму количеств никеля, кобальта, железа, хрома, титана, алюминия. Мешает молибден.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения 4,0 часа, включая время отбора пробы воздуха 2 минуты.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутным капельным электродом с записью полярограмм в переменно-токовом режиме, ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня песчаная.
Ступка фарфоровая, ГОСТ 9147-80Е.
Тигли кварцевые, ГОСТ 19908-80.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50 и 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 10, 15 и 25 мл.
Воронки химические стеклянные, ГОСТ 25336-82Е.
Палочки стеклянные, ГОСТ 25336-82Е.
Реактивы, растворы, материалы
Натрий вольфрамовокислый, 2-водный, ГОСТ 18289-78, чда.
Калия пиросульфат, ГОСТ 7172-76, чда.
Натрия гидроксид, ГОСТ 4328-77, хч, 5% раствор.
Калий хлорноватокислый (хлорат калия), ГОСТ 4235-77, хч, 0,5 М раствор.
Кислота серная, ГОСТ 4204-77, хч, 1 М раствор.
Кислота миндальная, ТУ 6-09-2943-77, ч, 0,1 М раствор (устойчив в течение недели при хранении в темном месте).
Стандартный раствор вольфрама N 1 с концентрацией 1,0 мг/мл готовят растворением 1,7940 г натрия вольфрамовокислого в дистиллированной воде в мерной колбе вместимостью 1000 мл. Раствор устойчив более года. Стандартные растворы N 2 и N 3 с концентрацией вольфрама 100 мкг/мл (устойчив в течение недели) и с концентрацией 5 мкг/мл (применяют свежеприготовленным) готовят соответствующим разбавлением стандартного раствора N 1 дистиллированной водой.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр типа АФА, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, помещенный в фильтродержатель. Для определения 1/2 ПДК достаточно отобрать 20 л воздуха. Пробы хранятся не менее месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2 часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ВОЛЬФРАМА
| N стандарта | Стандартный раствор N 3, мл | Вода дистиллированная, мл | Содержание вольфрама в градуировочном растворе, мкг |
| 1 | 0 | 2 | 0 |
| 2 | 0,1 | 1,9 | 0,5 |
| 3 | 0,2 | 1,8 | 1,0 |
| 4 | 0,5 | 1,5 | 2,5 |
| 5 | 1,0 | 1,0 | 5,0 |
| 6 | 1,2 | 0,8 | 6,0 |
| 7 | 1,6 | 0,4 | 8,0 |
Во все пробирки шкалы добавляют по 5,0 мл 1 М раствора серной кислоты, по 2,0 мл 0,5 М раствора хлората калия, по 1,0 мл 0,1 М раствора миндальной кислоты. Раствор переносят в электролизер и продувают аргоном в течение 5 — 7 минут, затем полярографируют.
Режим полярографирования переменно-токовый (на примере ПУ-Г), трехэлектродный ТАСТ-режим; поляризующее напряжение от -0,4 до -0,1 В; амплитуда квадратно-волновой формы 12 мВ; период капания 3 — 4 с; задержка 2,8 с; поляризация катодная; диапазон тока 0,5 x 100; координаты самописца: X = 2 x 100 мВ/см; Y = 1 x 100 мкА/см. Высоту пика вольфрама измеряют при потенциале E1/2 = -0,6 В.
Строят градуировочный график зависимости высоты пика вольфрама (мм) от его содержания в растворе (мкг).
Проверка градуировочного графика проводится 1 раз в 2 недели или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в кварцевый тигель, озоляют в муфельной печи при постепенном повышении температуры до 500 град. C. Зольный остаток выдерживают в печи при температуре 600 град. C около 20 мин. для получения оксида вольфрама (VI). По охлаждении остаток в тигле обрабатывают 2 мл 10% раствора гидроксида натрия и нагревают до полного растворения остатка. Раствор количественно переносят в мерную колбу вместимостью 50 мл и доводят до метки водой. Для анализа отбирают 0,5 мл полученного раствора, приливают 1,5 мл воды и далее обрабатывают и полярографируют аналогично градуировочным растворам.
Содержание вольфрама в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику или методом добавок.
Концентрацию вольфрама в воздухе (мг/куб. м) рассчитывают по формулам 1 или 4, 5 (Приложения 4.2, 4.3).
Измерение концентрации молибдена
Характеристика метода
Метод основан на восстановлении ионов молибдена на ртутно-капельном электроде на фоне смеси растворов серной кислоты, хлората калия и миндальной кислоты.
Потенциал восстановления молибдена относительно донной ртути равен 0,52 В.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения в полярографируемом растворе составляет 0,01 мкг/мл.
Нижний предел измерения в воздухе 0,5 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,5 до 25 мг/куб. м.
Определению молибдена мешают титан и вольфрам.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения около 2,5 часа, включая отбор пробы 2 минуты.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутно-капельным электродом с записью полярограмм в переменно-токовом режиме, ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Щипцы тигельные.
Баня песчаная.
Ступка фарфоровая, ГОСТ 9147-80Е.
Тигли фарфоровые, ГОСТ 9147-80Е.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 и 1000 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 мл.
Палочки стеклянные, ГОСТ 25336-82Е.
Воронки стеклянные, ГОСТ 25336-82Е, вместимостью 50 мл.
Реактивы, растворы, материалы
Молибдат аммония, 4-водный, ГОСТ 3765-78, хч.
Калия хлорат (хлорноватокислый), ГОСТ 4235-77, хч, 0,5 М раствор.
Серная кислота, ГОСТ 4294-77, концентрированная, 1,0 М и 10% растворы.
Азотная кислота, ГОСТ 4461-77, хч.
Калий азотнокислый, ГОСТ 4217-77, хч.
Натрий углекислый, ГОСТ 83-79, хч.
Миндальная кислота, ТУ 6-09-8943-77, хч, 0,5 М раствор (устойчив неделю при хранении в темном месте).
Плавень. Готовят растиранием в ступке двух частей натрия углекислого и одной части калия азотнокислого (по массе).
Стандартный раствор молибдена N 1 с концентрацией 1 мг/мл. Готовят растворением 1,8450 г молибдата аммония в воде в мерной колбе вместимостью 1000 мл. Раствор устойчив в течение года.
Стандартный раствор N 2 с концентрацией молибдена 10 мкг/мл готовят путем соответствующего разбавления раствора N 1. Раствор применяют свежеприготовленным.
Стандартный раствор N 3 с концентрацией молибдена 1 мкг/мл готовят разбавлением раствора N 2 в 10 раз, свежеприготовленный.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр типа АФА, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА. Для измерения 1/2 ПДК молибдена достаточно отобрать 20 л воздуха. Отобранные пробы хранятся не менее 2-х недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 45 минут) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МОЛИБДЕНА
| N стандарта | Стандартный раствор N 3, мл | Вода дистиллированная, мл | Содержание молибдена в градуировочном растворе, мкг |
| 1 | 0 | 2 | 0 |
| 2 | 0,1 | 1,9 | 0,1 |
| 3 | 0,2 | 1,8 | 0,2 |
| 4 | 0,4 | 1,6 | 0,4 |
| 5 | 0,6 | 1,4 | 0,6 |
| 6 | 0,8 | 1,2 | 0,8 |
| 7 | 1,0 | 1,0 | 1,0 |
Во все пробирки шкалы прибавляют по 5 мл 1 М раствора серной кислоты, затем по 2 мл 0,5 М раствора хлората калия и по 1 мл 0,5 М раствора миндальной кислоты.
Через 30 мин. градуировочные растворы заливают в электролизер, продувают аргоном и полярографируют.
Режим полярографирования переменно-токовый (на примере ПУ-1), двухэлектродный ТАСТ-режим; поляризующее напряжение от -0,4 В до -0,7 В; амплитуда квадратно-волновой формы 12 мВ; задержка 3,8 с; скорость развертки 9 мВ/с; поляризация катодная; диапазон тока 1 x 10; координаты самописца: X = 2 x 100 мВ/см; Y = 1 x 100 мкА/см.
Высоту пика молибдена измеряют при E1/2 = -0,52 В.
Строят градуировочный график зависимости высоты пика молибдена от его содержания в растворе (мкг). Проверка градуировочного графика проводится 1 раз в 2 недели или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с отобранной пробой переносят в фарфоровый тигель и добавляют по 0,2 мл концентрированных серной и азотной кислот. Содержимое тигля выпаривают на песчаной бане. Далее фильтр с пробой озоляют в муфельной печи, закрыв тигель крышкой, в течение 1 часа при постепенном повышении температуры до 500 град. C.
Тигель постепенно охлаждают, золу тщательно смешивают с 0,5 г плавня, помещают в муфельную печь, охлажденную до 300 град. C, повышают температуру до 800 град. C и оставляют на 30 мин. до полного сплавления смеси. Затем плав растворяют при нагревании в 5 мл 10% раствора серной кислоты и упаривают до влажных солей. Остаток растворяют в 5 мл 10% серной кислоты, раствор количественно переносят в мерную колбу вместимостью 100 мл, смывая тигель водой, и доводят объем до метки водой. На анализ отбирают в пробирки 0,2 — 2,0 мл раствора пробы, доводят объем до 2,0 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам.
Содержание молибдена в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику или методом добавок.
Концентрацию молибдена в воздухе (мг/куб. м) рассчитывают по формулам 1 или 4, 5 (Приложения 4.2, 4.3).
Измерение концентрации титана
Характеристика метода
Метод основан на восстановлении ионов титана на ртутно-капельном электроде на фоне серной кислоты в присутствии хлората калия и щавелевой кислоты.
Потенциал восстановления титана относительно хлоридсеребряного электрода — 0,2 В.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерений титана в полярографируемом растворе составляет 0,05 мкг/мл.
Нижний предел измерения в воздухе 1,3 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 1,3 до 63 мг/куб. м.
Определению мешает молибден.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерений 3 часа, включая отбор пробы воздуха 2 минуты.
Приборы, аппаратура, посуда
Полярограф ПУ-1 или другой системы с ртутным электродом (с записью полярограмм в переменно-токовом и постоянно-токовом режимах), ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Печь муфельная, МП-2УМ.
Горелка газовая.
Тигли платиновые, ГОСТ 6563-75, или кварцевые, ГОСТ 19908-80.
Щипцы тигельные с платиновыми наконечниками.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100, 500 и 1000 мл.
Пипетки мерные, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мм.
Воронки стеклянные, ГОСТ 25336-82Е.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 мл.
Пробирки с пришлифованными пробками, ГОСТ 10515-75.
Реактивы, растворы, материалы
Титана двуокись, ТУ 6-09-2166-77, чда.
Калия пиросульфат, ГОСТ 7172-76, чда.
Серная кислота, ГОСТ 4204-77, хч, концентрированная, 1 М и 0,5 М растворы.
Щавелевая кислота, 2-водная, ГОСТ 22180-76, хч, 0,5 М раствор.
Калий хлорноватокислый (хлорат), ГОСТ 4235-77, хч, 0,5 М раствор.
Стандартный раствор титана N 1 с концентрацией 100 мкг/мл. 0,1668 г высушенной до постоянного веса двуокиси титана сплавляют на газовой горелке в платиновом или кварцевом тигле с 3 г пиросульфата калия до получения прозрачного плава. После остывания плав растворяют при нагревании в 0,5 М серной кислоте, переносят в мерную колбу вместимостью 1000 мл и доводят объем до метки 0,5 М раствором серной кислоты. Раствор устойчив в течение 1 месяца.
Стандартный раствор N 2 с концентрацией титана 10 мкг/мл готовят разбавлением в 10 раз раствора N 1 водой. Раствор устойчив в течение суток.
Стандартный раствор N 3 с концентрацией титана 1 мкг/мл готовят разбавлением раствора N 2 водой. Применяют свежеприготовленным.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне, ГОСТ 10157-79.
Фильтр типа АФА, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА. Для измерения 1/2 ПДК титана достаточно отобрать 20 л воздуха. Пробы хранятся 2 недели.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ТИТАНА
| N стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Дистиллированная вода, мл | Содержание титана в градуировочном растворе, мкг |
| 1 | 2 | 3 | 4 | 5 |
| 1 | 0 | — | 1,0 | 0 |
| 2 | 0,5 | — | 0,5 | 0,5 |
| 3 | — | 0,1 | 0,9 | 1,0 |
| 4 | — | 0,2 | 0,8 | 2,0 |
| 5 | — | 0,3 | 0,7 | 3,0 |
| 6 | — | 0,4 | 0,6 | 4,0 |
| 7 | — | 0,5 | 0,5 | 5,0 |
Во все пробирки шкалы добавляют по 5 мл 1 М раствора серной кислоты, по 2 мл 0,5 М раствора хлората калия, по 2 мл 0,5 М раствора щавелевой кислоты. После добавления каждого реагента содержимое пробирок тщательно перемешивают. Перед проведением полярографирования градуировочные растворы выдерживают не менее 15 минут.
Градуировочные растворы в порядке возрастания концентрации заливают в электролизер, продувают в течение 5 мин. аргоном, затем полярографируют.
Режим полярографирования переменно-токовый (на примере ПУ-1), поляризующее напряжение от +0,1 В до -0,6 В; амплитуда квадратно-волновой формы 30 мВ; задержка 5,8 с; скорость развертки 5 мВ/с; поляризация катодная; диапазон тока 1 x 1; координаты самописца: X = 2 x 100 мВ/см; Y = 1 x 100 мкА/см.
Высоту пика титана измеряют при E1/2 = -0,2 В.
Строят градуировочный график зависимости высоты пика титана (мм) от его содержания в растворе (мкг). Проверка градуировочного графика проводится 1 раз в неделю или в случае использования новой партии реактивов.
Проведение измерения
Фильтр с пробой озоляют в платиновом или кварцевом тигле при 500 град. C и сплавляют с 0,5 г пиросульфата калия на газовой горелке до получения прозрачного плава. После сплавления плав растворяют в 0,5 М растворе серной кислоты при нагревании, раствор количественно переносят в мерную колбу вместимостью 50 мл и доводят объем до метки этим раствором.
0,2 — 1,0 мл раствора переносят в пробирку, доводят объем до 1,0 мл водой и далее обрабатывают и полярографируют аналогично градуировочным растворам.
Содержание титана в анализируемом объеме раствора пробы (мкг) определяют по градуировочному графику или методом добавок.
Концентрацию титана в воздухе (мг/куб. м) рассчитывают по формулам 1 или 4, 5 (Приложения 4.2, 4.3).
Измерение концентрации свинца, олова, кадмия и меди
Характеристика метода
Определение основано на восстановлении ионов свинца, олова, меди и кадмия на ртутно-капельном электроде на фоне 2% раствора щавелевой кислоты.
Потенциал восстановления относительно донной ртути меди — 0,36 В, свинца — 0,68 В, олова — 0,74 В, кадмия — 0,84 В.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения в полярографируемом растворе составляет для свинца 0,01 мкг/мл, олова 0,05 мкг/мл, меди 0,003 мкг/мл и кадмия 0,01 мкг/мл.
Нижний предел измерения в воздухе составляет для свинца и кадмия 0,005 мг/куб. м, для олова 0,025 мг/куб. м, для меди 0,002 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе свинца и кадмия от 0,005 до 0,5 мг/куб. м, олова от 0,025 до 1,0 мг/куб. м, меди от 0,002 до 0,005 мг/куб. м.
Измерению не мешает цинк.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения около 2 часов, включая время отбора проб 2 минуты.
Приборы, аппаратура, посуда
Полярограф ПУ-1, ППТ-1 или другой системы с ртутно-капельным электродом с записью полярограмм в переменно-токовом режиме, ГОСТ 22261-76.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня водяная.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 50, 100 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 и 100 мл.
Воронки стеклянные, ГОСТ 25336-82Е.
Реактивы, растворы, материалы
Все растворы готовят на бидистиллированной воде.
Свинец азотнокислый, ГОСТ 4236-77, хч, перекристаллизованный из воды и высушенный до постоянного веса при температуре 110 град. C.
Олово четыреххлористое, 5-водное, ТУ 6-09-3084-76.
Медь сернокислая, 5-водная, ГОСТ 4165-78, хч.
Кадмий азотнокислый: 4-водный, ГОСТ 6262-79, хч.
Азотная кислота, ГОСТ 4461-77, хч, разбавленная 1:1.
Щавелевая кислота, ГОСТ 22180-76, осч, 2% раствор.
Стандартный раствор свинца N 1 с концентрацией 1 мг/мл готовят растворением 0,1600 г азотнокислого свинца в воде в мерной колбе вместимостью 100 мл. Устойчив в течение 6 месяцев.
Стандартный раствор свинца N 2 с концентрацией 100 мкг/мл готовят путем разбавления стандартного раствора N 1 в 10 раз водой в мерной колбе. Устойчив 2 недели.
Стандартный раствор свинца N 3 с концентрацией 1 мкг/мл готовят соответствующим разбавлением раствора N 2 2% щавелевой кислоты в мерной колбе и применяют свежеприготовленным.
Стандартный раствор олова N 1 с концентрацией 1 мкг/мл готовят растворением 0,2965 г четыреххлористого олова в воде в мерной колбе вместимостью 100 мл. Устойчив 6 месяцев.
Стандартный раствор олова N 2 с концентрацией 100 мкг/мл готовят соответствующим разбавлением раствора N 1 водой в мерной колбе. Устойчив в течение 2 недель.
Стандартные растворы олова N 3 и 4 с концентрацией 10 и 1 мкг/мл готовят разбавлением раствора N 2 2% раствором щавелевой кислоты и применяют свежеприготовленными.
Стандартный раствор меди N 1 с концентрацией 1 мкг/мл готовят путем растворения 3,9293 г сернокислой меди, 5-водной, в воде в мерной колбе вместимостью 1 л. Устойчив 6 месяцев.
Стандартный раствор меди N 2 с концентрацией 100 мкг/мл готовят разбавлением раствора N 1 водой в 10 раз.
Стандартные растворы меди N 3 и N 4 с концентрацией 1,0 и 0,1 мкг/мл готовят разбавлением раствора N 2 2% раствором щавелевой кислоты в мерных колбах. Растворы не хранятся.
Стандартный раствор кадмия N 1 с концентрацией 1 мг/мл готовят растворением 0,2744 г азотнокислого кадмия в воде в мерной колбе вместимостью 100 мл. Устойчив в течение 6 месяцев.
Стандартный раствор кадмия N 2 с концентрацией 100 мкг/мл готовят разбавлением раствора N 1 водой в 10 раз в мерной колбе. Устойчив в течение 2 недель.
Стандартный раствор кадмия N 3 с концентрацией 1 мкг/мл готовят разбавлением раствора N 2 в 100 раз 2% раствором щавелевой кислоты и применяют свежеприготовленным.
Ртуть металлическая, ГОСТ 4658-73, хч.
Аргон в баллоне с редуктором, ГОСТ 10157-79, или азот в баллоне, ГОСТ 9293-74, очищенный от примеси кислорода.
Фильтр АФА-ХП-20, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА-ХП-20, укрепленный в фильтродержателе. Для определения 1/2 ПДК достаточно отобрать 20 л воздуха. Проба хранится не менее 2 недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение 3 часов) готовят согласно таблицам 1 — 4.
Таблица 1
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ СВИНЦА
| Номер стандарта | Стандартный раствор N 3, мл | Щавелевая кислота 2% раствор, мл | Концентрация свинца в градуировочном растворе, мкг/мл |
| 1 | 0 | 10,0 | 0 |
| 2 | 0,1 | 9,9 | 0,01 |
| 3 | 0,3 | 9,7 | 0,03 |
| 4 | 0,6 | 9,4 | 0,06 |
| 5 | 1,0 | 9,0 | 0,10 |
| 6 | 2,0 | 8,0 | 0,20 |
| 7 | 5,0 | 5,0 | 0,50 |
| 8 | 10,0 | — | 1,00 |
Таблица 2
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОЛОВА
| Номер стандарта | Стандартный раствор N 4, мл | Стандартный раствор N 3, мл | Щавелевая кислота 2% раствор, мл | Концентрация олова в градуировочном растворе, мкг/мл |
| 1 | 0 | — | 10,0 | 0 |
| 2 | 0,5 | — | 9,5 | 0,05 |
| 3 | 1,0 | — | 9,0 | 0,10 |
| 4 | 2,0 | — | 8,0 | 0,20 |
| 5 | 5,0 | — | 5,0 | 0,50 |
| 6 | 10,0 | — | 0 | 1,00 |
| 7 | — | 1,5 | 8,5 | 1,50 |
| 8 | — | 2,0 | 8,0 | 2,00 |
Таблица 3
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ МЕДИ
| Номер стандарта | Стандартный раствор N 4, мл | Щавелевая кислота 2% раствор, мл | Концентрация меди в градуировочном растворе, мкг/мл |
| 1 | 0 | 10,0 | 0 |
| 2 | 0,3 | 9,7 | 0,003 |
| 3 | 0,4 | 9,6 | 0,004 |
| 4 | 0,5 | 9,5 | 0,005 |
| 5 | 0,6 | 9,4 | 0,006 |
| 6 | 0,8 | 9,2 | 0,008 |
| 7 | 1,0 | 9,0 | 0,010 |
Таблица 4
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ КАДМИЯ
| Номер стандарта | Стандартный раствор N 3, мл | Щавелевая кислота 2% раствор, мл | Концентрация кадмия в градуировочном растворе, мкг/мл |
| 1 | 0 | 10,0 | 0 |
| 2 | 0,1 | 9,9 | 0,01 |
| 3 | 0,2 | 9,8 | 0,02 |
| 4 | 0,5 | 9,5 | 0,05 |
| 5 | 1,0 | 9,0 | 0,10 |
| 6 | 2,0 | 8,0 | 0,20 |
| 7 | 5,0 | 5,0 | 0,50 |
| 8 | 10,0 | 0 | 1,00 |
Градуировочные растворы последовательно заливают в электролизер, продувают в течение 5 минут аргоном или азотом и полярографируют.
Режим полярографирования на полярографе ППТ-1: переменно-токовый, поляризующее напряжение от -0,275 В до 0,9 В, скорость развертки 4 мВ/сек., амплитуда 4 мВ, период капания 3 — 4 сек., диапазон тока 0,5 — 40 (x 100).
Режим полярографирования на приборе ПУ-1: переменно-токовый, трехэлектродный ТАСТ-режим; поляризующее напряжение от -0,1 до -1,0 В; амплитуда квадратно-волновой формы 50 мВ; задержка 4,1 с; скорость развертки 2 мВ/с; поляризация катодная; координаты самописца: X = 2 x 100 мВ/см; Y = 5 x 10 мкА/см.
Высоту пика меди измеряют при E1/2 = -0,36 В, свинца — 0,68 В, олова — 0,74 В, кадмия — 0,84 В.
Строят градуировочные графики зависимости высот пиков (мм) свинца, олова, меди и кадмия от концентрации их в растворе (мкг/мл).
Проверка градуировочных графиков проводится 1 раз в 2 недели или в случае использования новой партии реактивов и растворов.
Проведение измерения
Фильтр с отобранной пробой переносят в химический стакан, заливают 10 мл азотной кислоты (1:1) и нагревают на кипящей водяной бане в течение 5 — 10 минут при перемешивании стеклянной палочкой. Фильтр отжимают на стенке стакана, промывают дважды 5 — 7 мл воды, промывные воды сливают в тот же стакан, после чего фильтр удаляют из стакана. Раствор выпаривают на кипящей водяной бане досуха. Для полного удаления паров азотной кислоты приливают 5 мл воды и упаривают вновь досуха. Сухой остаток растворяют в 10 мл 2% щавелевой кислоты. Раствор переносят в электролизер, продувают в течение 5 минут аргоном или азотом и полярографируют аналогично градуировочным растворам.
Концентрацию свинца, олова, меди и кадмия в полярографируемом растворе пробы (мкг/мл) определяют по соответствующим градуировочным графикам или методом добавок.
Концентрацию металлов в воздухе (мг/куб. м) рассчитывают по формулам 3 или 4, 5 (Приложения 4.2 и 4.3)
3.3. ИОНОМЕТРИЧЕСКИЕ МЕТОДЫ
Измерение концентраций борной кислоты и борного ангидрида
Характеристика метода
Определение основано на измерении потенциала тетрафторборатного электрода на фоне фосфатного буферного раствора с pH 5,4 после переведения борат-ионов в тетрафторборат-ионы по реакции с фтористоводородной кислотой.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения борной кислоты в растворе — 0,1 мкг/мл.
Нижний предел измерения в воздухе: борной кислоты — 0,3 мг/куб. м, борного ангидрида — 0,2 мг/куб. м (при отборе 40 л воздуха).
Диапазон измеряемых концентраций борной кислоты от 0,3 до 250 мг/куб. м, борного ангидрида — от 0,2 до 143 мг/куб. м.
Определению не мешают фосфаты, фториды, карбонаты, 100-кратные количества ионов меди, цинка, никеля, марганца, алюминия.
Суммарная погрешность измерения не превышает +/- 15%.
Время выполнения измерения 45 мин., включая отбор проб 4 мин.
Приборы, аппаратура, посуда
pH-метр типа pH-121, pH-340 или иономер ЭВ-74.
Электрод индикаторный тетрафторборатный, ЭМ-BF4-01, ТУ 25-05-2721-80.
Электрод сравнения, хлоридсеребряный ЭЛВ-1 МЗ, ГОСТ 16286-70.
Мешалка магнитная.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 100 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы полиэтиленовые, вместимостью 25 и 50 мл.
Бутыли полиэтиленовые, ТУ 6-19-45-74, вместимостью 100 и 1000 мл.
Секундомер, ГОСТ 5072-79.
Баня водяная.
Реактивы, растворы, материалы
Кислота борная, ГОСТ 9658-75, хч.
Натрия тетрафторборат, ТУ 6-09-1460-76, хч, 0,1 М раствор.
Кислота фтористоводородная, ГОСТ 1048-73, хч.
Аммиак водный, ГОСТ 3760-64, хч.
Калий фосфорнокислый однозамещенный, ГОСТ 4198-75, чда, 0,067 М раствор. Готовят растворением 9,078 г соли в воде в мерной колбе вместимостью 1 л.
Натрий фосфорнокислый двузамещенный двуводный, ГОСТ 245-76, чда, 0,067 М раствор. Готовят растворением 11,876 г соли в воде в мерной колбе вместимостью 1 л.
Буферный раствор с pH 5,4. В мерную колбу вместимостью 1 л вносят 31 мл раствора фосфорнокислого двузамещенного натрия и доводят объем до метки раствором фосфорнокислого однозамещенного калия. Раствор хранят в полиэтиленовой бутыли. Раствор устойчив в течение 2-х месяцев.
Стандартный раствор борной кислоты N 1 с концентрацией 1 мг/мл готовят растворением 0,1000 г борной кислоты в дистиллированной воде в мерной колбе вместимостью 100 мл. Основной стандартный раствор следует хранить в полиэтиленовой бутыли. Раствор устойчив в течение 2 месяцев.
Стандартный раствор N 2 с концентрацией тетрафторборат-ионов, соответствующей концентрации борной кислоты 100 мкг/мл, получают следующим образом: в полиэтиленовый стакан вносят 2,5 мл стандартного раствора борной кислоты N 1 с концентрацией 1 мг/мл и 7,5 мл дистиллированной воды. К раствору прибавляют 0,2 мл концентрированной фтористоводородной кислоты и выдерживают на водяной бане при температуре 50 — 60 град. C в течение 30 мин. После охлаждения в стакан приливают 0,1 мл концентрированного раствора аммиака, количественно переносят содержимое стакана в мерную колбу вместимостью 25 мл и перемешивают. Раствор устойчив в течение 2-х недель при хранении в полиэтиленовой бутыли.
Стандартный раствор N 3 с концентрацией тетрафторборат-ионов, соответствующей содержанию борной кислоты 10 мкг/мл, готовят разбавлением в 10 раз стандартного раствора N 2 водой. Устойчив в течение 2 дней.
Фильтр АФА-ХП-20, АФА-ВП-20, АФА-ХА-20, ТУ 95.743-80.
Отбор пробы воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, закрепленный в фильтродержателе. Для измерения 1/2 ПДК следует отобрать 40 л воздуха. Пробы хранятся в течение не менее двух недель.
Подготовка к измерению
Новый тетрафторборатный электрод вымачивают в 0,1 М растворе тетрафторбората натрия в течение 24 часов и затем 2 часа в дистиллированной воде. Далее электрод отмывают до потенциала ~ 250 мВ. Перед измерениями проверяют крутизну характеристики электрода (S). Если ![]() , электрод пригоден к работе.
, электрод пригоден к работе.
Градуировочные растворы (устойчивы в течение одного дня) готовят согласно таблице в мерных колбах вместимостью 25 мл.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ БОРНОЙ КИСЛОТЫ
| N стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Концентрация борной кислоты в градуировочном растворе, мкг/мл | |
| 1 | — | — | 0 | 0 |
| 2 | 0,25 | — | 0,1 | 5,793 |
| 3 | 1,00 | — | 0,4 | 5,190 |
| 4 | — | 0,5 | 2,0 | 4,490 |
| 5 | — | 1,0 | 4,0 | 4,190 |
| 6 | — | 2,5 | 10,0 | 3,793 |
| 7 | — | 10,0 | 40,0 | 3,190 |
| 8 | — | 20,0 | 80,0 | 2,889 |
Во все колбы шкалы прибавляют по 5 мл фосфатного буферного раствора, доводят объем до метки водой и перемешивают.
Полученные растворы последовательно в порядке возрастания концентраций переносят в полиэтиленовый стакан, вместимостью 50 мл, отпускают в него измерительный электрод, хлоридсеребряный электрод сравнения, якорь магнитной мешалки и измеряют величину электродного потенциала при постоянном перемешивании магнитной мешалкой. Показания снимают через 3 минуты.
Строят градуировочный график зависимости потенциала электрода от отрицательного логарифма концентрации тетрафторборат-ионов (![]() ), выраженной в г-ион/л. Проверку градуировочного графика проводят по нескольким точкам в день анализа проб.
), выраженной в г-ион/л. Проверку градуировочного графика проводят по нескольким точкам в день анализа проб.
Проведение измерения
Фильтр переносят в полиэтиленовый стакан вместимостью 25 — 50 мл и отмывают 4 — 5 раз горячей (70 град. C) дистиллированной водой порциями по 5 мл при перемешивании стеклянной палочкой. Фильтр отжимают на стенке стакана, сливая раствор в мерную колбу вместимостью 25 мл. После охлаждения раствора до комнатной температуры доводят содержимое колбы до метки водой и перемешивают.
5 мл раствора пробы помещают в полиэтиленовый стакан вместимостью 25 мл, прибавляют 5 мл воды и 0,2 мл концентрированной фтористоводородной кислоты. Раствор выдерживают на водяной бане с температурой 50 — 60 град. C в течение 30 минут, после чего пробу охлаждают до комнатной температуры и приливают 0,1 мл концентрированного раствора аммиака. Содержимое стакана количественно переносят в мерную колбу вместимостью 25 мл, приливают 5 мл фосфатного раствора, доводят объем до метки водой и перемешивают.
Полученный раствор возвращают в полиэтиленовый стакан и измеряют величину электродного потенциала аналогично градуировочным растворам.
Содержание борной кислоты в анализируемом объеме раствора пробы в г-ион/л определяют по градуировочному графику.
Расчет концентрации
Концентрацию борной кислоты (C) в воздухе (мг/куб. м) вычисляют по формуле:
![]() ,
,
где:
а — anti(![]() ) — антилогарифм концентрации тетрафторборат-ионов, выраженной в г-ион/л;
) — антилогарифм концентрации тетрафторборат-ионов, выраженной в г-ион/л;
86,8 x 1000 — коэффициент пересчета концентрации тетрафторборат-иона из г-ион/л в мкг/мл;
0,71 — коэффициент пересчета концентрации тетрафторборат-иона на концентрацию борной кислоты;
25 — общий объем раствора пробы, мл;
5 — объем пробы, взятый для анализа, мл;
![]() — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л. Коэффициент пересчета борной кислоты на борный ангидрид — 0,57.
— объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л. Коэффициент пересчета борной кислоты на борный ангидрид — 0,57.
Измерение концентрации фтористого водорода и солей фтористоводородной кислоты
Характеристика метода
Определение основано на измерении потенциала фторидного электрода на фоне 0,1 М раствора цитрата натрия с pH 5,2 — 5,6.
Отбор проб проводится с концентрированием на фильтр типа АФА, помещенный в фильтродержатель (для улавливания фторидов), и соединенную с ним сорбционную трубку (для улавливания фтористого водорода) (метод 1) или с использованием системы двух последовательно соединенных фильтродержателей, в один из которых помещен фильтр АФА (для улавливания фторидов), а во второй — беззольный фильтр «белая лента», импрегнированный двузамещенным фосфатом калия (для улавливания фтористого водорода) (метод 2).
Нижний предел измерения концентрации фтор-ионов в растворе 0,04 мкг/мл.
Нижний предел измерения концентрации в воздухе фтористого водорода 0,1 мг/куб. м, хорошо растворимых фторидов 0,25 мг/куб. м, плохо растворимых фторидов 1,0 мг/куб. м (при отборе 10 л воздуха).
Диапазоны измеряемых концентраций фтористого водорода от 0,1 до 5,0 мг/куб. м, хорошо растворимых фторидов от 0,25 до 12,5 мг/куб. м, плохо растворимых от 1,0 до 50,0 мг/куб. м.
Определению не мешают вещества, присутствующие в воздухе при процессах сварки.
Суммарная погрешность измерения не превышает +/- 15%.
Время выполнения измерения 40 минут, включая отбор пробы 3 мин.
Приборы, аппаратура, посуда
pH-метр типа pH-121, pH-340 или иономер ЭВ-74.
Мешалка магнитная ММ-2 или аналогичная.
Электрод фторидный, ЭF-У1, ТУ 48-1301-61-78.
Электрод хлоридсеребряный, ЭВЛ-1МЗ, ГОСТ 16286-70.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77, с двумя конусами.
Печь муфельная, МП-2УМ.
Тигли платиновые с крышками, ГОСТ 6563-75.
Щипцы тигельные.
Груша резиновая.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50, 100 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Стаканы химические, ГОСТ 25336-80Е, вместимостью 50 и 100 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25 и 50 мл.
Бутыли полиэтиленовые, ТУ 6-19-45-74, вместимостью 100, 250 и 500 мл.
Мензурка полиэтиленовая, вместимостью 30 мл (аптечная).
Воронка стеклянная, ГОСТ 25336-82Е.
Сорбционная трубка стеклянная с насадкой из стеклянной крошки, СТ 212 или 312, ТУ 25-1110.039.20-84.
Полиэтиленовая Г-образная трубка, диаметром 7 — 10 мм, длиной 7 — 10 см.
Реактивы, растворы, материалы
Натрий фтористый, ГОСТ 4463-76, хч, перекристаллизованный из воды с осаждением этиловым спиртом и высушенный до постоянного веса при температуре 105 — 110 град. C, 0,1 М, 0,01 М, 0,001 М, 0,0001 М, 0,00001 М растворы. Для получения 0,1 М раствора 4,1990 г фтористого натрия растворяют в дистиллированной воде в мерной колбе вместимостью 1 л. Путем соответствующего разбавления водой 0,1 М раствора получают серию растворов меньшей концентрации. Растворы хранят в полиэтиленовых бутылях.
Натрий углекислый безводный, ГОСТ 83-79, хч.
Борная кислота, ГОСТ 9656-75, хч.
Натрий лимоннокислый, трехзамещенный (цитрат натрия), ГОСТ 22280-76, чда.
Этиловый спирт-ректификат, ГОСТ 5963-67 или ГОСТ 18300-72, перегнанный.
Кислота соляная, ГОСТ 3118-77, хч.
Глицерин, ГОСТ 6259-75, чда.
Калий углекислый безводный, ГОСТ 83-79, хч.
Калий фосфорнокислый двузамещенный, ГОСТ 2493-75, хч, чда, 10% водный раствор, хранят в полиэтиленовой емкости.
Буферный раствор с pH 5,2 — 5,6. Готовят растворением 35,76 г цитрата натрия в мерной колбе вместимостью 1 л в 500 мл дистиллированной воды. После растворения соли в колбу приливают 6,2 мл концентрированной соляной кислоты, 100 мл этилового спирта, доводят раствор до метки водой и тщательно перемешивают. Раствор устойчив в течение 2-х недель.
Поглотительный раствор для обработки сорбционных трубок. В мерную колбу вместимостью 50 мл вносят 1,5 г карбоната калия, 7,5 мл глицерина, доводят раствор до метки водой и перемешивают. Раствор устойчив в течение 2-х месяцев.
Основной раствор фторида натрия с концентрацией фтор-ионов 100 мкг/мл готовят растворением 0,2210 г фторида натрия в дистиллированной воде в мерной колбе вместимостью 1000 мл.
Раствор хранят в полиэтиленовой бутыли. Устойчив в течение 2-х недель.
Стандартный раствор с концентрацией фтор-ионов 10 мкг/мл готовят соответствующим разбавлением основного раствора буферным раствором. Раствор устойчив в течение недели.
Фильтры типа АФА.
Фильтры обеззоленные «синяя лента» и «белая лента», ТУ 6-09-1678-77.
Отбор пробы воздуха
Метод 1
Подготовка сорбционных трубок к отбору проб. Слой стеклянных гранул в трубке смачивают с помощью пипетки 0,2 мл поглотительного раствора карбоната калия и глицерина. Раствор распределяют по поверхности гранул с помощью резиновой груши, надетой на выходной конец трубки. Трубки закрывают заглушками и помещают в полиэтиленовый мешок. Срок хранения подготовленных трубок 2 недели.
Воздух с объемным расходом 3 л/мин. аспирируют через фильтр типа АФА, помещенный в фильтродержатель, и соединенную с ним с помощью Г-образной полиэтиленовой трубки сорбционную трубку. Во время отбора пробы трубка устанавливается вертикально.
Для измерения 1/2 ПДК фтористого водорода и солей фтористоводородной кислоты следует отобрать 10 л воздуха.
Пробы сохраняются в течение 2-х недель.
Метод 2
Подготовка фильтров для отбора проб.
Из фильтров «белая лента» вырезают кружки диаметром, равным диаметру фильтродержателя, и погружают их в 10% раствор фосфорнокислого калия, помещенного в полиэтиленовую емкость. После смачивания фильтры вынимают пинцетом и помещают в вертикальном положении в полиэтиленовые мензурки по нескольку штук для высыхания при комнатной температуре. Высушенные фильтры хранят в полиэтиленовой емкости с закрытой крышкой. Для выбора фильтров, содержащих минимальное количество фтора, следует из разных партий фильтров «белая лента» выбрать по 5 штук и после вышеуказанной обработки их 10% раствором фосфорнокислого калия провести анализ раствора по методике, описанной ниже. Партию фильтров, в которой обнаружено больше 1 мкг фтор-ионов, бракуют.
Воздух с объемным расходом 3 — 5 л/мин. аспирируют через систему двух последовательно соединенных фильтродержателей, в один из которых помещен фильтр АФА, а в другой — бумажный фильтр «белая лента», пропитанный раствором фосфорнокислого калия и высушенный.
Для измерения 1/2 ПДК фтористого водорода и солей фтористоводородной кислоты достаточно отобрать 10 л воздуха. Пробы хранятся в герметичной полиэтиленовой емкости или полиэтиленовом пакете в течение 1 месяца.
Подготовка к измерению
Подготовка фторидного электрода. Новый фторидный электрод выдерживают в 0,001 М растворе фтористого натрия в течение 24 часов и затем 2 часа в дистиллированной воде. Далее электрод отмывают до потенциала E = 300 — 330 мВ.
Проверяют крутизну характеристики электрода, для чего измеряют его потенциал в двух растворах фтористого натрия 0,0001 М и 0,00001 М и рассчитывают крутизну характеристики электрода по формуле:
![]() ,
,
где:
![]() и
и ![]() — значения потенциалов для первого и второго растворов фтористого натрия, мВ;
— значения потенциалов для первого и второго растворов фтористого натрия, мВ;
![]() и
и ![]() — отрицательные логарифмы концентрации ионов фтора первого и второго растворов.
— отрицательные логарифмы концентрации ионов фтора первого и второго растворов.
Если ![]() , электрод пригоден к работе.
, электрод пригоден к работе.
Градуировочные растворы (устойчивы в течение 2-х часов) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ФТОРА
| N стандарта | Стандартный раствор N 2, мл | Отрицательный логарифм концентрации фтора (pF) | Концентрация фтора в градуировочном растворе, мкг/мл |
| 1 | 2 | 3 | 4 |
| 1 | 0 | — | 0 |
| 2 | 0,1 | 5,66 | 0,04 |
| 3 | 0,2 | 5,38 | 0,08 |
| 4 | 0,25 | 5,26 | 0,1 |
| 5 | 0,75 | 4,8 | 0,3 |
| 6 | 1,25 | 4,58 | 0,5 |
| 7 | 2,5 | 4,28 | 1,0 |
| 8 | 3,75 | 4,10 | 1,5 |
| 9 | 5,0 | 3,96 | 2,0 |
Градуировочные растворы готовят согласно таблице в мерных колбах вместимостью 25 мл, доводя объем в каждой колбе до метки буферным раствором. Измеряют потенциал фторидного электрода в градуировочных растворах, относительно хлоридосеребряного электрода сравнения, используя для измерения стеклянный стакан вместимостью 50 мл. Измеряют потенциал фторидного электрода при постоянном перемешивании раствора магнитной мешалкой до установления постоянного значения (10 — 3 минуты).
Измерения проводят в порядке возрастания концентрации фтор-иона. Перед измерением потенциала в каждом градуировочном растворе электроды осушают фильтровальной бумагой, обязательно помещают в контрольный раствор (раствор N 1 шкалы стандартов) и выдерживают в нем до установления первоначального значения потенциала фторидного электрода в этом растворе.
При выдерживании фторидного электрода в контрольном растворе до установления первоначального потенциала целесообразно использовать отдельный якорь магнитной мешалки («вертушку»).
После проведения измерений электроды оставляют в растворе, соответствующем средней точке шкалы стандартов.
Строят градуировочный график зависимости электродного потенциала (E, мВ) от отрицательного логарифма концентрации фтор-иона, выраженной в г-ион/л (pF). Проверка градуировочного графика проводится каждые 2 — 3 дня или в случае использования новой партии реактивов.
Проведение измерения
Определение концентрации фтористого водорода
Метод 1
Сорбционную трубку протирают фильтровальной бумагой и помещают в высокий стакан вместимостью 100 мл входным отверстием вниз. Пленку поглотительного раствора смывают 25 мл буферного раствора, приливая его с помощью пипетки в трубку. Тщательно перемешивают раствор в стакане путем многократного прокачивания его через сорбционную трубку с помощью резиновой груши, надетой на выходной конец трубки. (Раствор не должен попадать в грушу!) Раствор переносят в измерительный стакан вместимостью 50 мл, опускают в него якорь магнитной мешалки, фторидный и хлоридсеребряный электроды и измеряют электродный потенциал после установления постоянного значения.
Перед измерением потенциала в растворе пробы якорь магнитной мешалки, фторидный и хлоридсеребряный электроды осушают фильтровальной бумагой, промывают контрольным буферным раствором и выдерживают в нем до установления первоначального значения, после чего вновь осушают и погружают их в анализируемый раствор.
Аналогично поступают после измерения каждого анализируемого раствора.
Метод 2
При отборе пробы воздуха на фильтр, импрегнированный фосфатом калия, фильтр помещают в полиэтиленовую мензурку, приливают 5 мл буферного раствора и перемешивают полиэтиленовой или стеклянной палочкой. Фильтр отжимают на стенке мензурки, раствор сливают в мерную колбу вместимостью 25 мл. Обработку фильтра повторяют еще 4 раза, каждый раз перенося смывы в ту же колбу. При этом степень десорбции фтор-ионов достигает 97%. Раствор в колбе доводят до метки буферным раствором, перемешивают, переносят в измерительный стакан. Измерение потенциала проводят так же, как указано выше.
Определение концентрации хорошо растворимых солей фтористоводородной кислоты
Фильтр АФА с отобранной пробой помещают в воронку с бумажным фильтром «синяя лента» и смывают дробными порциями буферным раствором в мерную колбу вместимостью 25 мл. Доводят объем раствора до метки буфером. 10 мл полученного раствора вносят в колбу вместимостью 25 мл, доводят объем до метки буферным раствором, перемешивают, переливают в электролитическую ячейку и измеряют потенциал фторидного электрода аналогично градуировочным растворам, как описано выше.
Определение концентрации плохо растворимых солей фтористоводородной кислоты
Фильтр АФА после обработки буферным раствором помещают в платиновый тигель, подсушивают и озоляют в муфельной печи при температуре 400 — 500 град. C. К остатку прибавляют 0,5 г карбоната натрия и 0,05 г борной кислоты, закрывают платиновой крышкой и сплавляют при 950 — 1000 град. C в течение 15 — 20 минут. После охлаждения плав растворяют в 20 мл воды, подкисленной 1 мл концентрированной соляной кислоты, при перемешивании на магнитной мешалке. Раствор количественно переносят в мерную колбу вместимостью 25 мл и доводят объем до метки водой (pH раствора 3 — 5).
2,5 мл полученного раствора плава вносят в мерную колбу вместимостью 25 мл и доводят до метки буферным раствором.
Раствор переносят в электролитическую ячейку и измеряют значение потенциала фтористого водорода, как описано выше.
Расчет концентраций фтористого водорода и солей фтористоводородной кислоты в воздухе
По градуировочному графику определяют соответствующий величине потенциала отрицательный логарифм концентрации фтор-иона (pF), выраженной в г-ион/л.
Концентрацию фтористого водорода в воздухе (C мг/куб. м) вычисляют по формуле:
![]() .
.
Концентрацию солей фтористоводородной кислоты в воздухе (по фтор-иону) вычисляют по формуле:
![]() ,
,
где:
а — anti(![]() ) — антилогарифм концентрации фтор-иона, выраженной в г-ион/л;
) — антилогарифм концентрации фтор-иона, выраженной в г-ион/л;
19 x ![]() — коэффициент пересчета концентрации фтор-иона из г-ион/л в мкг/мл;
— коэффициент пересчета концентрации фтор-иона из г-ион/л в мкг/мл;
в — объем раствора пробы, взятый для анализа, мл;
25 — общий объем раствора пробы, мл;
![]() — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
— объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
3.4. АТОМНО-АБСОРБЦИОННЫЕ МЕТОДЫ
Измерение концентрации кобальта, никеля, меди, цинка, кадмия, свинца, железа, марганца, молибдена, олова, вольфрама, оксида ванадия и оксидов хрома
Характеристика метода
Метод основан на измерении абсорбции атомами исследуемого элемента резонансного излучения с соответствующей длиной волны. Атомизация осуществляется в пламени.
Отбор проб производится с концентрированием на фильтр.
Нижние пределы измерения металлов в растворе, в воздухе и диапазоны измеряемых концентраций в воздухе представлены в таблице 1.
Таблица 1
| Элемент | Оптимальный рабочий диапазон концентраций металла в фотометрируемом объеме раствора, мкг/мл | Нижний предел измерения концентрации металла в воздухе, мг/м3 | Объем отобранного воздуха, л | Диапазон измеряемых концентраций в воздухе, мг/м3 |
| 1 | 2 | 3 | 4 | 5 |
| Кобальт | 0,1 — 20 | 0,01 | 100 | 0,01 — 2,0 |
| Никель | 0,05 — 5,0 | 0,005 | 100 | 0,005 — 0,5 |
| Медь | 0,2 — 50 | 0,02 | 100 | 0,02 — 5,0 |
| Цинк | 0,1 — 50 | 0,01 | 100 | 0,01 — 5,0 |
| Марганец | 0,2 — 30 | 0,02 | 100 | 0,02 — 3,0 |
| Кадмий | 0,2 — 20 | 0,02 | 100 | 0,02 — 2,0 |
| Свинец | 0,2 — 20 | 0,007 | 300 | 0,007 — 0,7 |
| Хром | 0,1 — 100 | 0,005 | 200 | 0,005 — 5,0 |
| Олово | 2 — 200 | 0,2 | 100 | 0,2 — 20 |
| Железо | 0,1 — 100 | 0,01 | 100 | 0,01 — 10,0 |
| Молибден | 5,0 — 200 | 0,5 | 100 | 0,5 — 20,0 |
| Ванадий | 1,5 — 150 | 0,05 | 300 | 0,05 — 5,0 |
| Вольфрам | 100 — 1500 | 3,3 | 300 | 3,3 — 50 |
Измерение концентрации каждого металла специфично в присутствии остальных при использовании моноэлементных ламп с полым катодом.
В присутствии относительно высоких концентраций железа из-за спектральных наложений последнее мешает определению ряда металлов, в частности кобальта и никеля. В этих случаях необходимо устанавливать концентрацию железа и вводить его в градуировочные растворы для определения указанных металлов в тех же количествах.
Суммарная погрешность измерений не превышает +/- 20%.
Время выполнения измерений (при условии определения всех элементов из одной пробы) 5 — 6 часов, включая отбор пробы 10 — 15 минут.
Аппаратура, приборы, посуда
Атомно-абсорбционный спектрофотометр любого типа с чувствительностью по определяемым элементам не ниже указанной в таблице 1.
Набор моноэлементных ламп с полым катодом в соответствии со списком определяемых элементов.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня песчаная, ТУ 46-775-77.
Тигли фарфоровые низкие N 3 и крышки к ним с ушками, ГОСТ 9447-78.
Стаканы химические, ГОСТ 1770-74, вместимостью 50 мл.
Пробирки химические, ГОСТ 25336-82, размером 15 x 150 мм.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 50, 100 и 1000 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Воронки стеклянные, ГОСТ 25336-82Е.
Флаконы и банки полиэтиленовые цилиндрические, ТУ 6-19-45-74, вместимостью 500 и 1000 мл.
Вся посуда обрабатывается в течение нескольких часов азотной кислотой, разбавленной 1:1, с последующим ополаскиванием бидистиллированной водой.
Реактивы, растворы, материалы
Все растворы готовят на бидистиллированной воде.
Аммоний ванадиевокислый, мета, ГОСТ 9336-75, чда.
Железо металлическое, восстановленное, ТУ 6-09-2227-72, хч.
Кадмия оксид, ГОСТ 11120-75, чда.
Кобальт (II) азотнокислый, 6-водный, ТУ 6-09-20-85-75, осч.
Марганец (II) сернокислый, 5-водный, ГОСТ 435-77, чда.
Медь (II) сернокислая, 5-водная, ГОСТ 4165-78, хч.
Никель двухлористый, 6-водный, ГОСТ 4038-79, хч.
Олово гранулированное, ТУ 6-09-2704-78, чда.
Свинец гранулированный, ТУ 6-09-3523-74, г.
Калий хромовокислый, ГОСТ 4458-75, хч.
Цинк гранулированный, ГОСТ 989-75, хч.
Молибдена (VI) оксид, ТУ 6-09-4471-77, хч.
Натрий вольфрамовокислый, 2-водный, ГОСТ 18289-78, чда.
Натрия гидроксид, ГОСТ 4328-77, хч, 2% и 10% растворы.
Спирт этиловый, ГОСТ 5962-67, ректификат.
Азотная кислота, ГОСТ 11125-73, осч, концентрированная, разбавленная 1:1 и 2 М раствор.
Соляная кислота, ГОСТ 14261-77, осч, концентрированная, 2 М раствор.
Хлорная кислота, ТУ 6-09-2878-73, хч.
Серная кислота, ГОСТ 4204-77, хч, 10% раствор.
Основные стандартные растворы с концентрацией каждого металла 1 мг/мл готовят из соответствующих металлов, их солей или оксидов (таблица 2) путем растворения взятых навесок в растворах кислот, щелочей или воде, переведения растворов в мерные колбы вместимостью 1 л и доведения объемов до метки водой. Растворы хранят в полиэтиленовых флаконах. Устойчивы в течение 6 месяцев.
Рабочие стандартные растворы с концентрацией металла 10 мкг/мл и менее готовят соответствующим разбавлением основного стандартного раствора водой непосредственно перед употреблением.
Ацетилен, ГОСТ 5457-75, в баллоне с редуктором.
Пропан, ГОСТ 5542-78, в баллоне с редуктором.
Закись азота, Государственная фармакопея СССР, статья 455, 10-е издание, 1968 г.
Фильтры АФА-ХА-20, ТУ 95-743-80.
Бумажные фильтры обеззоленные, ТУ 6-09-1678-77, «белая лента», «красная лента».
Таблица 2
ПРИГОТОВЛЕНИЕ ОСНОВНЫХ СТАНДАРТНЫХ РАСТВОРОВ
| Элемент | Исходный реагент | Навеска, г | Растворитель | Концентрация металла, мг/мл |
| 1 | 2 | 3 | 4 | 5 |
| Кобальт | Кобальт азотнокислый, 6-водный | 4,9386 | Вода | 1,0 |
| Никель | Никель двухлористый, 6-водный | 4,0490 | -«- | 1,0 |
| Медь | Медь (II) сернокислая, 5-водная | 3,9295 | -«- | 1,0 |
| Ванадий | Аммоний ванадиевокислый, мета | 2,2964 | -«- | 1,0 |
| Хром | Калий хромовокислый | 3,7346 | -«- | 1,0 |
| Марганец | Марганец сернокислый, 5-водный | 4,3881 | 10% раствор серной кислоты | 1,0 |
| Цинк | Цинк гранулированный | 1,0000 | 50 мл 5 М раствора соляной кислоты | 1,0 |
| Кадмий | Кадмия оксид | 1,1422 | 20 мл 5 М раствора соляной кислоты | 1,0 |
| Свинец | Свинец гранулированный | 1,0000 | 50 мл 2 М раствора азотной кислоты | 1,0 |
| Олово | Олово гранулированное | 1,0000 | 200 мл концентрированной соляной кислоты и 5 мл концентрированной азотной кислоты | 1,0 |
| Железо | Железо металлическое | 1,0000 | 20 мл 5 М раствора соляной кислоты и 5 мл концентрированной азотной кислоты | 1,0 |
| Вольфрам | Натрий вольфрамовокислый, 2-водный | 1,7942 | 100 мл 2% раствора гидроксида натрия | 1,0 |
| Молибден | Молибдена оксид (VI) | 1,5003 | 10 мл 10% раствора гидроксида натрия | 1,0 |
Отбор проб воздуха
Воздух с объемным расходом 10 — 20 л/мин. аспирируют через фильтр АФА-ХА-20. Для измерения 1/2 ПДК металла следует отобрать 100 — 300 л воздуха (таблица 1). Отобранные пробы сохраняются в течение 2-х недель. Пробы, содержащие оксид хрома (VI), не хранятся.
Подготовка к измерению
Для построения градуировочных графиков готовят градуировочные растворы для определения каждого металла (не менее 4 — 5 концентраций), исходя из диапазона измеряемых концентраций (таблица 1) с использованием стандартных растворов с концентрацией 10 мкг/мл.
Градуировочные растворы подают в атомизатор и измеряют величину светопоглощения при соответствующей длине волны, указанной в таблице 3. Аналогично измеряют аналитический сигнал контрольного раствора. Условия фотометрирования представлены в таблице 3. Насадки для горелок выбирают в соответствии с типом пламени.
Таблица 3
УСЛОВИЯ СПЕКТРОФОТОМЕТРИРОВАНИЯ
| Элемент | Длина волны, нм | Ширина щели, мм | Тип пламени |
| Кобальт | 240,7 | 0,2 | Пропан — воздух |
| Никель | 232,0 | 0,2 | -«- |
| Медь | 324,7 | 0,7 | -«- |
| Марганец | 279,5 | 0,7 | -«- |
| Цинк | 213,9 | 0,7 | -«- |
| Кадмий | 228,8 | 0,2 | -«- |
| Свинец | 283,3 | 0,7 | -«- |
| Хром | 357,9 | 0,7 | Ацетилен — воздух |
| Железо | 248,3 | 0,2 | -«- |
| Молибден | 313,3 | 0,7 | -«- |
| Ванадий | 318,5 | 0,7 | Ацетилен — закись азота |
| Олово | 235,5 | 0,2 | -«- |
| Вольфрам | 255,1 | 0,2 | -«- |
Проводят пять параллельных измерений каждой концентрации. Для каждого металла строят градуировочный график зависимости величины светопоглощения растворов от концентрации металла в растворе (мкг/мл). Проверку градуировочных графиков по нескольким точкам осуществляют в день анализа проб.
Проведение измерения
При наличии хрома (VI) фильтр с отобранной пробой помещают на стеклянную воронку, смачивают 2 — 3 каплями этилового спирта и обрабатывают 10 мл теплой воды. Фильтрат подают в атомизатор и анализируют на содержание хрома (VI). Для определения остальных металлов подсушенный фильтр переносят в тигель и обрабатывают 1 мл концентрированной соляной кислоты и 0,5 мл концентрированной азотной кислоты.
Затем к раствору добавляют 1 мл хлорной кислоты. Тигель ставят на песчаную баню и медленно упаривают до влажных солей. Охлажденный остаток растворяют в 10 мл воды. Раствор при необходимости фильтруют, подают в атомизатор и фотометрируют аналогично градуировочным растворам. Одновременно с пробами аналогичным образом обрабатывают 3 — 5 чистых фильтров той же партии (контрольные пробы).
Концентрацию металла в мкг/мл в анализируемой пробе находят по градуировочным графикам.
Концентрацию металла в воздухе (мг/куб. м) рассчитывают по формуле 3 (Приложение 4.2).
Коэффициент пересчета хрома на оксид хрома (VI) — 1,92.
Коэффициент пересчета хрома на оксид хрома (III) — 1,46.
Коэффициент пересчета ванадия на оксид ванадия (V) — 1,58.
Измерение концентрации оксида алюминия
Характеристика метода
Метод основан на измерении абсорбции атомами алюминия резонансного излучения с длиной волны 309,3 нм. Атомизация осуществляется в пламени ацетилен — закись азота.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения содержания алюминия в растворе 1 мкг/мл.
Нижний предел измерения оксида алюминия в воздухе 1 мг/куб. м (при отборе 50 л воздуха).
Диапазон измеряемых концентраций оксида алюминия в воздухе от 1 до 100 мг/куб. м.
Определению не мешают 10-кратные количества марганца, 50-кратные количества хрома, свинца, никеля, титана, стронция, кремния, железа, цинка, бария, фтора.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерения, включая отбор проб, 1,5 часа.
Приборы, аппаратура, посуда
Атомно-абсорбционный спектрофотометр с атомизацией в пламени.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Тигли платиновые, ГОСТ 6563-75.
Щипцы тигельные с платиновыми наконечниками.
Печь муфельная, МУ-2УМ.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50 и 100 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Пробирки химические, ГОСТ 10515-75, размером 15 x 150 мм.
Реактивы, растворы, материалы
Алюминий гранулированный, ТУ 6-09-3742-74, чда.
Калия пиросульфат (калий сернокислый пиро), ГОСТ 7172-76, чда.
Кислота соляная, ГОСТ 3118-77, хч, разбавленная 1:1.
Стандартный раствор N 1 с концентрацией алюминия 1 мг/мл готовят растворением 0,1000 г алюминия в 20 мл соляной кислоты (1:1). Раствор количественно переводят в мерную колбу вместимостью 100 мл и доводят до метки водой.
Стандартный раствор N 2 с концентрацией алюминия 100 мкг/мл (устойчив в течение недели) готовят соответствующим разбавлением стандартного раствора N 1 дистиллированной водой.
Ацетилен, ГОСТ 5457-75, в баллоне с редуктором.
Закись азота, Гос. фармакопея СССР, 1968, статья 455, 10-е издание.
Фильтры АФА-ХП, АФА-ВП и АФА-ХА, ТУ 95.743-80.
Отбор проб воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр АФА, помещенный в фильтродержатель. Для измерения 1/2 ПДК достаточно отобрать 50 л воздуха. Отобранные пробы хранятся в течение месяца.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение рабочего дня) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ АЛЮМИНИЯ
| Номер стандарта | Ст. раствор N 1, мл | Дистиллированная вода, мл | Концентрация алюминия в градуировочном растворе, мкг/мл |
| 1 | 0,0 | 10,0 | 0,0 |
| 2 | 0,1 | 9,9 | 1,0 |
| 3 | 0,2 | 9,8 | 2,0 |
| 4 | 0,5 | 9,5 | 5,0 |
| 5 | 1,0 | 9,0 | 10,0 |
| 6 | 2,0 | 8,0 | 20,0 |
| 7 | 5,0 | 5,0 | 50,0 |
| 8 | 10,0 | 0,0 | 100,0 |
Градуировочные растворы подают в распылительную камеру прибора, где происходит разложение растворов в атомные пары, и измеряют поглощение излучения при длине волны 309,3 нм.
Условия фотометрирования:
Длина волны — 309,3 нм.
Ширина щели — 0,2 мм.
ФЭУ — 39 А.
Постоянная времени — 1 или 5.
Поддиапазон измерений 2:1 или 5:1.
Сигнал — 3.
Расход ацетилена — 700 л/ч.
Расход закиси азота — 400 л/ч.
Строят градуировочный график зависимости значения светопоглощения растворов от концентрации алюминия (мкг/мл), проверку которого проводят по нескольким точкам в день анализа проб.
Проведение измерения
Фильтр с отобранной пробой помещают в платиновый тигель и озоляют при температуре 500 град. C. После охлаждения в тигель вносят 0,5 г пиросульфата калия, хорошо перемешивают и сплавляют, постепенно повышая температуру до 650 — 700 град. C, выдерживают при данной температуре 10 мин. По охлаждении плав растворяют в горячей воде и количественно переносят в мерную колбу вместимостью 25 мл. Анализируемый раствор пробы подают в атомизатор прибора и фотометрируют аналогично градуировочным растворам. Подобным образом анализируют «холостую» пробу.
Концентрацию алюминия в мкг/мл в анализируемом растворе находят по градуировочному графику.
Концентрацию оксида алюминия в воздухе (мг/куб. м) рассчитывают по формуле 3 (Приложение 4.2).
Коэффициент пересчета алюминия на оксид алюминия — 1,88.
Измерение концентрации оксида кальция
Характеристика метода
Метод основан на измерении абсорбции атомами кальция резонансного излучения с длиной волны 429,7 нм. Атомизация осуществляется в пламени.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения содержания оксида кальция в растворе 0,5 мкг/мл.
Нижний предел измерения в воздухе 0,25 мг/куб. м (при отборе 20 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,25 до 12,5 мг/куб. м.
Определению не мешают 10-кратные количества марганца, 25-кратные количества хрома и цинка, 50-кратные количества железа, 100-кратные количества никеля.
Мешает титан, даже следы. Мешающее влияние титана устраняется проведением атомизации пробы в пламени закись азота — ацетилен.
Суммарная погрешность измерения не превышает +/- 20%.
Время выполнения измерений 30 минут, включая отбор пробы 10 минут.
Приборы, аппаратура, посуда
Атомно-абсорбционный спектрофотометр с атомизацией в пламени.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 25, 50 и 100 мл.
Стаканы химические, ГОСТ 25336-82Е, вместимостью 50 мл.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Пробирки химические, ГОСТ 10515-75, размером 15 x 150 мм.
Реактивы, растворы, материалы
Кальция оксид, ГОСТ 8677-66, хч.
Спирт этиловый, ГОСТ 18300-72.
Кислота азотная, ГОСТ 4461-77, хч, 5% раствор.
Стандартный раствор N 1 оксида кальция с концентрацией 1 мг/мл готовят растворением 0,1000 г оксида кальция в 5% растворе азотной кислоты в мерной колбе вместимостью 100 мл. Раствор устойчив в течение месяца.
Стандартные растворы N 2 с концентрацией оксида кальция 100 мкг/мл (устойчив в течение недели) и N 3 с концентрацией 10 мкг/мл (устойчив в течение рабочего дня) готовят путем соответствующего разбавления стандартного раствора N 1 5% раствором азотной кислоты.
Ацетилен, ГОСТ 5457-75, в баллоне с редуктором.
Фильтр типа АФА, ТУ 95.743-80.
Отбор проб воздуха
Воздух с объемным расходом 10 л/мин. аспирируют через фильтр типа АФА, помещенный в фильтродержатель. Для измерения 1/2 ОБУВ, установленного для аэрозолей щелочей (ориентировочный), достаточно отобрать 20 л воздуха.
Отобранные пробы хранятся в течение 2 недель.
Подготовка к измерению
Градуировочные растворы (устойчивы в течение суток) готовят согласно таблице.
Таблица
ШКАЛА ГРАДУИРОВОЧНЫХ РАСТВОРОВ ДЛЯ ОПРЕДЕЛЕНИЯ ОКСИДА КАЛЬЦИЯ
| Номер стандарта | Стандартный раствор N 3, мл | Стандартный раствор N 2, мл | Азотная кислота, 5% раствор, мл | Концентрация оксида кальция в градуировочном растворе, мкг/мл |
| 1 | 0 | 0 | 10,0 | 0 |
| 2 | 0,5 | — | 9,5 | 0,5 |
| 3 | 1,0 | — | 9,0 | 1,0 |
| 4 | — | 0,5 | 9,5 | 5,0 |
| 5 | — | 1,0 | 9,0 | 10,0 |
| 6 | — | 2,5 | 7,5 | 25,0 |
Градуировочные растворы подают в распылительную камеру прибора, где происходит разложение растворов в атомные пары, и измеряют поглощение излучения при длине волны 422,7 нм.
Условия фотометрирования:
Длина волны — 422,7 нм.
Ширина щели — 0,1 мм.
ФЭУ — 39 А.
Постоянная времени — 1.
Поддиапазон измерений — 1:1.
Сигнал — 2.
Расход ацетилена — 130 л/ч.
Расход воздуха — 680 л/ч.
Строят градуировочный график зависимости величины светопоглощения растворов от концентрации оксида кальция в мкг/мл, проверку которого проводят по нескольким точкам в день анализа проб.
Проведение измерения
Фильтр с отобранной пробой помещают в стакан, смачивают 2 — 3 каплями этанола и обрабатывают 10 мл 5% раствора азотной кислоты. Выдерживают при перемешивании в течение 10 минут, затем фильтр отжимают и раствор переливают в пробирку. При необходимости раствор фильтруют через фильтр «синяя лента».
Анализируемый раствор пробы подают в атомизатор прибора и фотометрируют аналогично градуировочным растворам.
Концентрацию оксида кальция в анализируемом растворе пробы в мкг/мл находят по градуировочному графику.
Концентрацию оксида кальция в воздухе (мг/куб. м) рассчитывают по формуле 3 (Приложение 4.2).
3.5. ГАЗОХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Измерение концентрации оксида углерода <*>
<*> Допускается измерение концентрации оксида углерода с помощью индикаторных трубок по ГОСТ 12.1.014-84.
Характеристика метода
Определение основано на использовании метода реакционной газовой хроматографии, включающего разделение оксида углерода и сопутствующих компонентов на колонке с молекулярными ситами, конверсию оксида углерода в метан и детектирование метана пламенно-ионизационным детектором.
Отбор проб проводится без концентрирования.
Нижний предел измерения в хроматографируемом объеме воздуха — 0,001 мкг.
Нижний предел измерения в воздухе 0,5 мг/куб. м.
Диапазон измеряемых концентраций оксида углерода в воздухе от 0,5 до 500 мг/куб. м.
Определению не мешают двуокись углерода, метан и другие углеводороды.
Суммарная погрешность измерения не превышает +/- 4%.
Время измерения 10 минут, включая отбор пробы.
Приборы, аппаратура, посуда
Хроматограф с пламенно-ионизационным детектором, ТУ 25-05.2.815-82.
Колонка хроматографическая из нержавеющей стали длиной 2 м и внутренним диаметром 3 мм.
Метанатор — трубка из нержавеющей стали длиной 25 см и внутренним диаметром 3 мм.
Аспирационное устройство.
Система газоснабжения СГС-2, ТУ 6-09-1.550.044-72.
Микрокомпрессор ВК-1, ТУ 25-06.926-77.
Печь муфельная электрическая, ТУ 16-931-098-67 или ПМ-2УМ.
Набор сит «Физприбор».
Секундомер, ГОСТ 5072-79.
Шприцы цельностеклянные, ТУ 64-1-1279-75, вместимостью 100 мл.
Пипетки газовые, ГОСТ 1770-74, вместимостью 250, 500 мл.
Реактивы, растворы, материалы
Натрия хлорид, ГОСТ 4233-77, хч, насыщенный раствор.
Молекулярные сита CaA, ТУ 38401-231-78.
Проволока нихромовая марки Х20Н80, ГОСТ 12766-67.
Газообразный воздух в баллоне с редуктором или от микрокомпрессора, ГОСТ 11882-73.
Газообразный азот (или гелий) в баллоне с редуктором, ГОСТ 9293-74.
Газообразный водород в баллоне с редуктором, ГОСТ 3022-80, или от системы газоснабжения.
Аттестованная смесь оксида углерода в баллоне с редуктором (выпускаются Балашихинским кислородным заводом).
Отбор проб воздуха
Отбор проб воздуха проводят в газовые пипетки или цельностеклянные шприцы. Десятикратный объем воздуха протягивают с помощью аспирационного устройства со скоростью 0,1 — 0,5 л/мин. Пипетки закрывают заглушками.
Срок хранения проб, отобранных в газовые пипетки, 2 недели.
Подготовка к измерению
Подготовка метанатора.
Нихромовую проволоку диаметром 0,15 — 0,24 мм нарезают кусочками до 3 мм, прокаливают в муфеле при температуре 1000 град. C в течение двух часов. Охлажденным катализатором механически заполняют метанатор, концы которого закрывают пробками из стекловаты. Метанатор, согнутый в виде U-образной трубки, помещают в испаритель, изолируют стекловатой и закрывают металлическим кожухом. Собирают газовую схему согласно рисунку и проводят активацию катализатора в течение 6 часов в токе водорода при температуре 325 град. C (нагрев метанатора обеспечивается термостатом испарителя).
Молекулярные сита CaA измельчают, отсеивают фракцию 0,25 — 0,5 мм и прокаливают при температуре 350 град. C в течение четырех часов. Охлажденным в эксикаторе адсорбентом механически заполняют колонку.
Перед началом работы адсорбент дополнительно активизируют пропусканием газа-носителя (при отсоединенном выходе колонки) при температуре 250 град. C в течение пяти часов.
Ввод пробы в хроматограф осуществляют краном — дозатором через дозирующую петлю объемом 2 мл. Для этого 20 мл воздуха отбирают из баллона с аттестованной смесью углерода и продувают дозирующую петлю через кран-дозатор.
Условия анализа:
Длина колонки 2 м
Диаметр колонки 3 мм
Наполнитель колонки молекулярные сита CaA,
фракция 0,25 — 0,5 мм
Температура колонки 80 град. C
Температура метанатора 325 град. C
Газ-носитель азот или гелий
Скорость потока газа-носителя 35 мл/мин.
Скорость потока водорода 35 мл/мин.
Скорость потока воздуха 270 мл/мин.
Скорость диаграммной ленты 10 мм/мин.
Объем анализируемой пробы 2 мл
Ориентировочное время удерживания CO 1 мин. 50 сек.
Проведение измерения
При определении оксида углерода в газовой фазе пробу воздуха из пипетки или шприца вытесняют в дозирующую петлю насыщенным раствором хлорида натрия.
Количественное определение оксида углерода проводят методом сравнения. Для этого перед анализом пробы и после анализа вводят в хроматограф аттестованную смесь оксида углерода аналогично пробам.
Концентрация оксида углерода и аттестованной смеси должна быть близка по величине к анализируемой.
Расчет концентрации
Концентрацию оксида углерода «C» (мг/куб. м) воздуха вычисляют по формуле:
![]() ,
,
где:
а — количество оксида углерода в объеме аттестованной смеси,
введенной в хроматограф, мкг;
![]() — высота пика оксида углерода в аттестованной смеси, мм;
— высота пика оксида углерода в аттестованной смеси, мм;
![]() — высота пика оксида углерода в пробе, мм;
— высота пика оксида углерода в пробе, мм;
v — объем дозы, мл;
![]() — коэффициент пересчета.
— коэффициент пересчета.
3.6. ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Измерение концентрации оксида кальция
Характеристика метода
Определение основано на осаждении кальция в виде оксалата, переведении его в раствор путем обработки серной кислотой и титровании оксалат-ионов раствором перманганата калия.
Отбор проб воздуха проводится с концентрированием на фильтр.
Нижний предел измерения оксида кальция в анализируемом объеме раствора 14,0 мкг.
Нижний предел измерения в воздухе 0,25 мг/куб. м (при отборе 280 л воздуха).
Диапазон измеряемых концентраций в воздухе от 0,25 до 5,0 мг/куб. м.
Определению не мешают железо, марганец, магний, алюминий.
Суммарная погрешность измерения не превышает +/- 25%.
Время выполнения измерения 2,5 часа, включая отбор проб 30 минут.
Приборы, аппаратура, посуда
Микробюретка, ГОСТ 1770-74Е, вместимостью 2 мл.
Центрифуга электрическая лабораторная.
Аспирационное устройство.
Фильтродержатель, ТУ 95.72.05-77.
Баня водяная.
Стаканы химические, ГОСТ 25336-82Е.
Пробирки центрифужные, ГОСТ 25336-82.
Пипетки, ГОСТ 20292-74Е, вместимостью 1, 2, 5 и 10 мл.
Цилиндры мерные, ГОСТ 1770-74Е, вместимостью 25, 50 и 100 мл.
Колбы мерные, ГОСТ 1770-74Е, вместимостью 100 и 1000 мл.
Реактивы, растворы, материалы
Кислота азотная, ГОСТ 4461-77, хч, 2% и 10% растворы.
Кислота серная, ГОСТ 4204-77, хч, 1 н. раствор.
Аммиак водный, ГОСТ 3760-64, хч, чда, концентрированный и 2% раствор.
Аммоний щавелевокислый, 1-водный, ГОСТ 5712-78, хч, насыщенный на холоду.
Спирт этиловый, ГОСТ 18300-72 или 5963-67.
Калий марганцевокислый, ТУ 6-09-2540-72, 0,01 н. раствор, готовят из фиксанала.
Фильтры типа АФА, ТУ 95-743-80.
Фильтры бумажные, обеззоленные, ТУ 6-09-1678-77, «синяя лента».
Бумага индикаторная, универсальная pH 1 — 10, ТУ 6-09-1881-71.
Отбор проб
Воздух с объемным расходом 15 — 20 л/мин. аспирируют через фильтр АФА, помещенный в фильтродержатель. Для измерения 1/2 ОБУВ, установленного для аэрозолей и щелочей (ориентировочный), следует отобрать 280 л воздуха. Отобранные пробы хранятся в течение 2 недель.
Проведение измерения
Фильтр с отобранной пробой помещают в стакан, смачивают 2 — 3 каплями спирта и обрабатывают 10 мл 10% раствора азотной кислоты. Оставляют на 10 — 15 минут, затем фильтр отжимают, а раствор фильтруют.
2 мл азотнокислого раствора пробы помещают в центрифужную пробирку и нейтрализуют сначала концентрированным раствором аммиака (0,2 — 0,25 мл), а затем 2% раствором аммиака до pH 5 — 6; если pH раствора будет щелочным, его снова нейтрализуют 2% раствором азотной кислоты (по универсальной индикаторной бумаге). После этого в пробирку приливают 1 мл насыщенного раствора щавелевокислого аммония, взбалтывают и оставляют на 30 минут. По истечении этого времени раствор центрифугируют в течение 5 минут, затем раствор сливают, внешние стенки пробирки вытирают фильтровальной бумагой, а внутреннюю смывают 5 мл 2% раствора аммиака и снова центрифугируют. Обработку повторяют 2 — 3 раза.
Отмытый осадок оксалата кальция растворяют в 2 мл 1 н. раствора серной кислоты. Пробирку помещают в водяную баню и нагревают до 70 — 75 град. C. Горячий раствор титруют в пробирке 0,01 н. раствором перманганата калия до розовой окраски, не исчезающей в течение 1 минуты. В таких же условиях ставят контрольный опыт с фильтром и реактивами.
Расчет концентрации
Концентрацию оксида кальция «C» в воздухе (мг/куб. м) вычисляют по формуле:
![]() ,
,
где:
![]() — количество раствора перманганата калия, пошедшее на титрование контрольного раствора, мл;
— количество раствора перманганата калия, пошедшее на титрование контрольного раствора, мл;
а — количество раствора перманганата калия, пошедшее на титрование пробы, мл;
в — общий объем раствора пробы, мл;
б — объем пробы, взятый на титрование, мл;
0,28 — коэффициент пересчета (1 мл 0,01 н. раствора перманганата калия соответствует 0,28 мг CaO);
![]() — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
— объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
Приложение 1
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВРЕДНЫХ ВЕЩЕСТВ, ВЫДЕЛЯЮЩИХСЯ В ВОЗДУХ ПРИ ПРОВЕДЕНИИ ДУГОВЫХ И ПЛАЗМЕННЫХ ПРОЦЕССОВ
| N | Вещество | Химическая формула | Молекулярная масса | Агрегатное состояние вещества | Плотность, г/см3 | Тпл., °C | Ткип., °C | Растворимость | Примечание | ||
| в воде | в кислотах | в щелочах | |||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 1 | Азота (II) оксид | NO | 30,0 | Газ без цвета и запаха | 1,34 х |
-163,6 | -151,8 | 0,047 (20 град.) | н.р. | н.р. | |
| 2 | Азота (IV) оксид | NO2 | 46,0 | Газ буро-красного цвета с характерным запахом | 3,3 х |
-10,2 | 22,4 | х.р. | — | — | |
| 3 | Алюминий | Al | 26,9 | Металл серебристо-белого цвета | 2,70 | 660 | 2500 | н.р. | р. | р. | При нагревании растворим в органических кислотах |
| 4 | Алюминия оксид | Al2O3 | 101,9 | Аморфный порошок белого цвета | 3,50 — 3,97 | 2050 | 2980 | н.р. | почти н.р. | почти н.р. | В растворимое состояние переводят путем сплавления со щелочами или пиросульфатом калия |
| 5 | Борный ангидрид | B2O3 | 69,6 | Бесцветные гигроскопические кристаллы | 1,78 — 1,85 | 557 | 1860 | х.р. | — | х.р. | |
| 6 | Борная кислота | H3BO3 | 61,8 | Бесцветные кристаллы | 1,43 — 1,49 | 185 | — | р. | — | х.р. | Растворяется в этаноле, глицерине |
| 7 | Ванадий | V | 50,9 | Металл серебристого цвета | 6,11 | 1715 | 3400 | н.р. | р. в азотной, плавиковой кислотах и царской водке | н.р. | |
| 8 | Ванадия оксид (V) (ванадиевый ангидрид) | V2O5 | Кристаллы красно-желтого или красного цвета | 3,36 | 670 | — | р. | х.р. | х.р. | ||
| 9 | Ванадия оксид (III) | V2O3 | 149,8 | Черный кристаллический порошок | 5,84 | 1970 | — | н.р. | сл.р. (х.р. в кипящей азотной кислоте, плавиковой кислоте) |
н.р. | |
| 10 | Вольфрам | W | 183,8 | Металл светло-серого цвета | 19,3 | 3410 | 5930 | — | х.р. в.р. смеси с плавиковой и азотной кислот | в кипящих щелочах | При прокаливании до 400 — 500 °C превращается в трехокись вольфрама |
| 11 | Вольфрама оксид (VI) | WO3 | 231,8 | Тяжелый порошок канареечного цвета | 7,16 | 1470 | — | н.р. | только в плавиковой кислоте | р. | Возгоняется при температурах выше 1750 °C |
| 12 | Железо | Fe | 55,8 | Металл серебристо-белого цвета | 7,87 | 1539 | 3200 | н.р. | х.р. пр. н.р. в конц. растворах серной кислоты | н.р. | Окисляется в присутствии влаги и кислорода |
| 13 | Железа оксид (III) | Fe2O3 | 159,7 | Кристаллы от темно-красного до черно-фиолетового цвета | 4,69 — 5,25 | 1562 | — | н.р. | х.р. | н.р. | |
| 14 | Кадмий | Cd | 112,4 | Металл серебристо-белого цвета | 8,65 | 321,03 | 767 | — | х.р. | — | В порошке загорается |
| 15 | Кадмия оксид | CdO | 128,3 | Кристаллы или аморфная масса коричневого цвета | 8,15 — крист. 6,95 — аморф. |
900 | — | сл.р. | х.р. | — | |
| 16 | Кальция оксид | CaO | 56,0 | Аморфный порошок белого цвета | 3,16 | 2580 | 2850 | х.р. | х.р. | — | |
| 17 | Кобальт | Co | 58,9 | Металл, похожий на сталь с синеватым отливом | 8,84 | 1492 | 3100 | н.р. | р. в разбавленной азотной и серной кислотах, в царской водке | — | |
| 18 | Кобальта оксид | CoO | 74,9 | Порошок оливково-зеленого цвета | 5,68 | 1935 | — | н.р. | л.р. | н.р. | При 2800° разлагается с потерей кислорода |
| 19 | Магний | Mg | 24,3 | Металл серебристо-белого цвета | 1,74 | 651 | 1107 | р. при нагревании | х.р. | — | Растворяется при нагревании в спирте |
| 20 | Магния оксид (магнезия) | MgO | 40,3 | Рыхлый порошок белого цвета | 3,5 — 3,9 | 2500 | 2800 | — | х.р. | — | |
| 21 | Марганец | Mn | 54,9 | Металл серебристо-белого цвета | 7,2 — 7,4 | 1244 | 2095 | — | х.р. | — | Растворим в уксусной кислоте |
| 22 | Марганца оксид | MnO | 70,9 | Кристаллы серо-зеленого цвета | 5,18 | 1650 | — | мало р. | р. | — | При 3400° возгоняется с диссоциацией |
| 23 | Марганца диоксид | MnO2 | 86,9 | Порошок от серого до серо-черного цвета | 5,026 | — | — | мало р. | х.р. | н.р. | Растворим в перекиси водорода, особенно хорошо в кислой среде |
| 24 | Медь | Cu | 63,5 | Металл красноватого цвета | 8,92 | 1083 | 2350 | н.р. | р. | н.р. | |
| 25 | Меди оксид (II) | CuO | 79,5 | Кристаллы или порошок черного цвета | 6,4 — 6,45 | — | — | н.р. | х.р. | — | |
| 26 | Молибден | Mo | 95,9 | Металл светло-серого цвета | 10,2 | 2630 | 4800 | н.р. | р. в царской водке, смеси азотной и плавиковой кислот, в концентрированной азотной и при нагревании в концентрированной серной кислотах | сл.р. | |
| 27 | Молибдена оксид (VI) | MoO3 | 143,9 | Бесцветные кристаллы с зеленоватым оттенком | 4,69 | 795 | 1155 | мало р. | р. в плавиковой и конц. серной кислотах | легко р. | Растворим в растворах аммиака и карбонатов |
| 28 | Никель | Ni | 58,7 | Металл | 8,90 | 1453 | 3000 | н.р. | х.р. | н.р. | |
| 29 | Никеля оксид | NiO | 74,7 | Кристаллы зеленого цвета | 7,45 | 1950 | — | н.р. | х.р. | н.р. | При прокаливании переходит в серо-черные кристаллы, трудно растворимые в кислотах |
| 30 | Озон | O3 | 48 | Газ голубоватого цвета с резким запахом, взрывоопасен | 2,145 х |
-192,7 | -111,9 | р. | р. в уксусной кислоте | — | Растворим в тетрахлорметане |
| 31 | Олово | Sn | 118,7 | Металл серого цвета с синеватым оттенком | 11,34 | 327,4 | 1740 | н.р. | х.р. в азотной, крепкой соляной кислотах | р. при кипячении | |
| 32 | Свинец | Pb | 207,2 | Металл серого цвета с синеватым оттенком | 11,34 | 327,4 | 1740 | сл.р. при доступе кислорода воздуха | х.р. в азотной кислоте, при доступе кислорода в уксусной и разбавленных минеральных | тр.р. | |
| 33 | Свинца оксид | PbO | 223,2 | Рыхлый порошок желтого или красного цвета | 9,35 и 9,63 | 886 | 1472 | почти н.р. | х.р. | р. | Тр.р. в растворах ацетата, тартрата аммония |
| 34 | Титан | Ti | 47,9 | Металл серебристо-белого цвета | 4,5 | 1670 | 3227 | н.р. | р. в горячей азотной, плавиковой и при нагревании в разбавленной соляной кислотах | н.р. | |
| 35 | Титана диоксид | TiO2 | 79,9 | Порошок белого цвета | 4,18 — 4,25 | 1850 | ок. 3000 | н.р. | н.р. | — | В раствор может быть переведен при длительном кипячении с концентрированной серной кислотой, взаимодействием с плавиковой кислотой или сплавлением с гидросульфатом натрия или калия |
| 36 | Углерода монооксид (угарный газ) | CO | 28,0 | Газ без цвета и запаха | — | -191,5 | -205 | тр.р. | — | — | |
| 37 | Фтористый водород | HF | 20,0 | Бесцветный газ | 0,9885 (12°) (жидкого) | -83,37 | 19,43 | х.р. | — | х.р. | |
| 38 | Фторид натрия | NaF | 42,0 | Бесцветные кристаллы | 2,79 | 992 | 1695 | 4,1 (20°) | — | — | Не растворим в спирте |
| 39 | Фторид калия | KF | 58,1 | Бесцветные кристаллы | 2,505 | 857 | 1500 | 96,0 (25°) | — | — | Расплывается на воздухе |
| 40 | Фторид лития | LiF | 25,9 | Бесцветные кристаллы | 2,635 (20°) | 870 | 1681 | 0,25 (20°) | х.р. азотной, серной и плавиковой кислотах | р. при нагревании | Не растворим в органических растворителях |
| 41 | Фторид алюминия | AlF3 | 84,0 | Бесцветные кристаллы | 3,10 | 1040 | 1256 | 0,56 (25°) | р. только в кипящей серной кислоте | н.р. | |
| 42 | Фторид кальция | CaF2 | 78,1 | Бесцветные кристаллы | 3,18 | 1360 | 2500 | 0,017 (20°) | — | — | |
| 43 | Криолит | Na3AlF6 | Бесцветные кристаллы | 2,95 — 3,01 | 1000 — 1009 | — | 0,035 (25°) | — | р. при кипячении | ||
| 44 | Хром | Cr | 52,00 | Металл серо-стального цвета, хрупкий | 7,19 | 1890 | 2480 | н.р. | х.р. в разбавленных растворах соляной и серной кислот, т.р. в конц. азотной и серной кислотах | н.р. | |
| 45 | Хрома оксид (III) | Cr2O3 | 152,0 | Кристаллы темно-зеленого цвета | 5,21 | 2275 | — | н.р. | тр.р. | н.р. | Легко переходит в раствор с раствором щелочного бромата при нагревании |
| 46 | Хрома оксид (VI) | CrO3 | 100,0 | Кристаллические иглы темно-красного цвета | 2,80 | 170 — 196 | — | х.р. | х.р. | х.р. | Сильный окислитель, легко восстанавливается до Cr (III). Не растворяется в уксусной кислоте |
| 47 | Цинк | Zn | 65,4 | Блестящий металл голубовато-серебристого цвета | 7,14 | 419,5 | 906 +/- 1 | н.р. | х.р. | р. | |
| 48 | Цинка оксид | ZnO | 81,4 | Порошок бесцветный или кристаллы гексогональной формы | 5,70 | — | — | тр.р. | х.р. | р. | T возг. 1800 °C. Растворим в аммиаке |
| 49 | Цирконий | Zr | 91,2 | Металл, похожий на сталь | 6,49 | 1857 | 3600 | н.р. | н.р. на холоде, р. в горячей 60% серной кислоте, царской водке, плавиковой кислоте | н.р. | |
| 50 | Циркония диоксид | 123,2 | Порошок белого цвета. Имеет несколько модификаций | 5,73 | 2900 | 4300 | н.р. | Слабопрокаленный р. в кислотах. Сильнопрокаленный — только в плавиковой и конц. серной кислотах | — | Может быть переведен в раствор путем сплавления со щелочами или карбонатами щелочных металлов |
Приложение 2
ПРЕДЕЛЬНО ДОПУСТИМЫЕ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ, ВЫДЕЛЯЮЩИХСЯ В ВОЗДУХ В ВИДЕ ТВЕРДОЙ И ГАЗОВОЙ СОСТАВЛЯЮЩИХ СВАРОЧНЫХ АЭРОЗОЛЕЙ
| NN пп | Вещество | ПДК в воздухе рабочей зоны (м.р./с.с <*>). мг/м3 | Класс опасности | Наименование нормативного документа |
| 1 | 2 | 3 | 4 | 5 |
| Твердая составляющая сварочного аэрозоля (ТССА) | ||||
| 1. | Алюминий, его сплавы, алюминия оксид (в том числе с примесью диоксида кремния) в виде аэрозоля конденсации | 2,0 | 4 | ГОСТ 12.1.005-76 «Воздух рабочей зоны» (Таблица 5) |
| 2. | Борная кислота | 10,0 | 4 | СН 245-71 |
| 3. | Бора оксид | 5,0 | 4 | ПДК вредных веществ в воздухе рабочей зоны N 841-70 от 30.04.70 |
| 4. | Ванадия (V) оксид (дым) | 0,1 | 1 | ГОСТ 12.1.005-76 (Таблица 4) |
| 5. | Ванадия (III) оксид | 0,5 | 2 | -«- |
| 6. | Вольфрам | 6,0 | 3 | -«- |
| 7. | Железо | — | — | — |
| 8. | Железа оксид | 6,0 | 4 | Ориентировочная ПДК |
| 9. | Кадмий и его неорганические соединения | 0,1/0,01 <*> | 1 | ПДК вредных веществ в воздухе рабочей зоны (Перечень N 27, N 4114-86) |
| 10. | Кальция оксид | — | — | — |
| 11. | Кобальт металлический, кобальта оксид | 0,5 | 2 | ГОСТ 12.1.005-76 (Таблица 4) |
| 12. | Кремния диоксид аморфный в смеси с оксидами марганца в виде аэрозоля конденсации с содержанием каждого из них более 10% | 1,0 | 3 | ГОСТ 12.1.005-76 (Таблица 5) |
| 13. | Магний | 1,0 | 3 | Ориентировочная |
| 14. | Магния оксид | 10,0 | 4 | Ориентировочная |
| 15. | Марганец | |||
| при его содержании в сварочном аэрозоле а) до 20% |
0,2 | 2 | ПДК. Перечень N 29, N 4280-87 | |
| б) от 20% до 30% | 0,1 | 2 | -«- | |
| 16. | Медь металлическая | 1/0,5 <*> | 2 | ГОСТ 12.1.005-76 (Таблица N 4) |
| 17. | Молибден металлический | 4/0,5 <*> | 2 | ПДК. Перечень N 23, N 2961-84 |
| 18. | Никель металлический, его оксиды (в пересчете на никель) | 0,05 | 1 | ПДК. Перечень N 14, N 2114-79 |
| 19. | Олово | 2,0 | — | Ориентировочная ПДК вредных веществ в воздухе и воде. Справочное пособие для выбора и гигиенической оценки методов обезвреживания промышленных отходов. Л.: Химия, 1975, с. 15 |
| 20. | Свинец и его неорганические соединения | 0,01/0,007 <*> | 1 | ГОСТ 12.1.005-76 (Таблица 4) |
| 21. | Титан, титана диоксид | 10,0 | 4 | ГОСТ 12.1.005-76 (Таблица 5) |
| 22. | Фтористоводородной кислоты соли а) хорошо растворимые |
1,0/0,2 <*> | ПДК. Перечень N | |
| б) плохо растворимые | 2,5/0,2 <*> | |||
| 23. | Хрома (III) оксид | 1,0 | 2 | ГОСТ 12.1.005-76 (Таблица 4) |
| 24. | Хрома (VI) оксид | 0,01 | 1 | -«- |
| 25. | Цинка оксид | 0,5 | 2 | ПДК. Перечень N 14, N 2114-79 |
| 26. | Цирконий металлический, циркония диоксид | 6,0 | 3 | ГОСТ 12.1.005-76 (Таблица 4) |
| Газовая составляющая сварочного аэрозоля (ГССА) | ||||
| 27. | Азота диоксид | 2,0 | 3 | ПДК. Перечень N 14, N 2114-79 |
| 28. | Озон | 0,1 | 1 | ГОСТ 12.1.005-76 (Таблица 4) |
| 29. | Углерода монооксид | 20,0 | 4 | -«- |
| 30. | Фтористый водород | 0,5/0,1 <*> | 2 | ПДК. Перечень N 31 |
<*> С.с. — средняя сменная концентрация.
Приложение 3
ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ ПРИ РАБОТЕ В ЛАБОРАТОРИИ С ВРЕДНЫМИ ВЕЩЕСТВАМИ И АППАРАТУРОЙ
Все сотрудники лабораторий, производящих химические и физико-химические исследования, должны соблюдать «Правила техники безопасности и производственной санитарии», разработанные и утвержденные МЗ СССР от 2 октября 1981 г. N 2455-81.
Ниже приведены основные положения этих «Правил».
1. К работе допускаются только лица, прошедшие инструктаж по технике безопасности (под расписку) в соответствии с правилами при работе в физико-химической лаборатории и инструкцией по противопожарной безопасности.
2. При работе с огнеопасными веществами (натрий, бензин, эфир и др.) надевается маска из органического стекла. В случае проведения указанных работ в комнате должно находиться не менее двух человек.
3. Переливание концентрированных кислот, растворов щелочей, аммиака и др. из больших бутылей в меньшие производится с помощью сифона и воронки. В качестве защитной одежды пользуются резиновыми перчатками и фартуками. Необходимо также надевать на лицо защитные очки или маску. Пролитые кислоты и щелочи следует осторожно нейтрализовать, большие количества засыпать песком.
4. Пересыпание едких щелочей производят в защитной маске или очках, резиновых перчатках и фартуке.
5. Ядовитые или причиняющие ожоги жидкости (растворы солей ртути, бензола, концентрированные кислоты, щелочи и др.) набираются только с помощью пипеток с насадками. Категорически запрещается производить отбор указанных жидкостей ртом.
6. При работе с легковоспламеняющимися веществами (эфир, бензол, петролейный эфир и др.) не должно быть по соседству огня и включенных электроплиток.
Нагревание этих веществ можно проводить на предварительно нагретой водяной или масляной бане. Проводить нагрев легковоспламеняющихся жидкостей на открытом огне и плитках открытого типа запрещается.
7. Общий запас одновременно хранящихся в каждом помещении легковоспламеняющихся жидкостей (ЛВЖ) не должен превышать суточного запаса, но быть не более 5 л.
8. Нагревание и перегонка более 1 л ЛВЖ производится только с разрешения заведующего лабораторией, в присутствии старшего научного сотрудника и с обязательной защитой лиц работающих масками.
9. В случае пролива ртути необходимо немедленно собрать ее механическим путем (с помощью резиновой груши, амальгамированной медной пластинки). Остаточное количество ртути обезвреживают 20-процентным раствором хлорного или 0,1-процентным подкисленным раствором марганцевокислого калия (1 г КМnО4 и 5 мл соляной кислоты в 1 л воды).
10. При ожогах кислотами и щелочами пораженный участок кожи сразу промывают большим количеством воды, затем на обожженное место накладывают примочку: при ожогах кислотой — из 2-процентного раствора соды, при ожогах щелочью — из слабого 1-процентного раствора уксусной кислоты.
11. При повреждении едкими веществами глаз их немедленно промывают большим количеством воды и затем 3-процентным раствором бикарбоната натрия (соды).
12. При термических ожогах 1 степени накладывают различные примочки: 96-процентный спирт или 2-процентный свежий раствор питьевой соды.
13. Для ликвидации случайно возникшего пожара пользуются только углекислотными огнетушителями, песком, асбестовыми одеялами. Последними также пользуются для тушения загоревшейся одежды.
14. При обнаружении запаха газа перекрывают общий кран вне лаборатории и сообщают об этом в аварийную службу.
15. При работе с физико-химической аппаратурой необходимо соблюдать правила работы с электротехническими устройствами с напряжением до 1000 В, опасным для жизни.
16. При работе с газовыми баллонами необходимо соблюдать требования ГОСТов 12.1.004-76. ССБТ «Пожарная безопасность. Общие требования» и 12.1.010-76. ССБТ «Взрывобезопасность. Общие требования».
Приложение 4
РАСЧЕТ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ
4.1. Приведение объема отобранного воздуха к стандартным условиям
Объем воздуха, аспирированного при отборе проб, приводят к стандартным условиям: температуре 20 град. C и барометрическому давлению 760 мм рт. ст. (101,33 кПа) по формуле:
![]() ,
,
где:
![]() — объем воздуха при температуре в месте отбора пробы, л;
— объем воздуха при температуре в месте отбора пробы, л;
P — атмосферное давление, мм рт. ст.;
t — температура воздуха в месте отбора пробы, град. C.
Для упрощения расчетов можно пользоваться коэффициентами К, вычисленными для температур в пределах от 6 до 40 град. C и давлений от 730 до 780 град. C (97,33 — 104,0 кПа), которые приведены во всех руководствах по анализу воздушной среды и в сборниках методических указаний на методы определения вредных веществ в воздухе.
4.2. Расчет результатов анализа с использованием градуировочных графиков
В тех случаях, когда градуировочный график для измерения концентрации вещества строят в координатах «величина аналитического сигнала — содержание вещества в градуировочном растворе (мкг)», концентрацию вещества в воздухе (C) вычисляют по формуле:
![]() , мг/куб. м, (1)
, мг/куб. м, (1)
где:
а — количество вещества, найденное в анализируемом объеме
раствора пробы по градуировочному графику, мкг;
б — объем раствора пробы, взятый на анализ, мл;
в — общий объем раствора пробы, мл;
![]() — объем отобранного на анализ воздуха, приведенный к стандартным условиям (температуре 20 град. C и атмосферному давлению 760 мм рт. ст. (101 кПа), л.
— объем отобранного на анализ воздуха, приведенный к стандартным условиям (температуре 20 град. C и атмосферному давлению 760 мм рт. ст. (101 кПа), л.
В случае, когда для анализа берут всю пробу, формула расчета упрощается:
![]() , мг/куб. м. (2)
, мг/куб. м. (2)
Если градуировочный график строят в координатах «величина аналитического сигнала — концентрация определяемого вещества в растворе (мкг/мл)», формула приобретает вид:
![]() , мг/куб. м, (3)
, мг/куб. м, (3)
где а — концентрация вещества в анализируемом растворе, мкг/мл.
4.3. Расчет результатов анализа с использованием метода добавки
Для расчета результатов полярографического анализа, наряду с методом градуировочного графика, используется метод добавок.
Для этого пробу, подготовленную для анализа (![]() ), полярографируют и вычисляют высоту полученного пика (
), полярографируют и вычисляют высоту полученного пика (![]() ). Затем в полярографическую ячейку добавляют небольшой объем (~ 0,5 мл) стандартного раствора определяемого металла (
). Затем в полярографическую ячейку добавляют небольшой объем (~ 0,5 мл) стандартного раствора определяемого металла (![]() ) с известной концентрацией (
) с известной концентрацией (![]() ) и снова снимают полярограмму, после чего вычисляют высоту полученного суммарного пика (
) и снова снимают полярограмму, после чего вычисляют высоту полученного суммарного пика (![]() ). Стандартный раствор следует добавлять в таком количестве, чтобы высота пика увеличилась в 1,5 — 2 раза в ходе записи полярограммы при том же диапазоне тока.
). Стандартный раствор следует добавлять в таком количестве, чтобы высота пика увеличилась в 1,5 — 2 раза в ходе записи полярограммы при том же диапазоне тока.
Концентрацию металла (![]() ) в исследуемом растворе (мкг/мл) рассчитывают по формуле:
) в исследуемом растворе (мкг/мл) рассчитывают по формуле:
![]() . (4)
. (4)
Концентрацию металла (C) в воздухе (мг/куб. м) вычисляют по формуле:
![]() , (5)
, (5)
где:
![]() — концентрация металла в исследуемом растворе, мкг/мл;
— концентрация металла в исследуемом растворе, мкг/мл;
![]() — общий объем исследуемого раствора пробы, мл;
— общий объем исследуемого раствора пробы, мл;
![]() — объем исследуемого раствора, взятого на анализ, мл;
— объем исследуемого раствора, взятого на анализ, мл;
![]() — объем раствора, подготовленного для полярографирования, мл;
— объем раствора, подготовленного для полярографирования, мл;
![]() — объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
— объем воздуха, отобранный для анализа и приведенный к стандартным условиям, л.
Приложение 5
РАСЧЕТ ПОГРЕШНОСТИ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИЙ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ
Расчет погрешности измерения проводится в соответствии с ГОСТ 12.1.016-79 с Изменениями N 1 (опубликовано: Государственные стандарты СССР. Информационный указатель, 1983, N 9, с. 237 — 240).
Приложение 6
ПРИМЕРНЫЙ ПЕРЕЧЕНЬ ОСНОВНЫХ ЭЛЕМЕНТОВ И СОЕДИНЕНИЙ, ВЫДЕЛЯЮЩИХСЯ В СОСТАВЕ ТССА И ГССА ПРИ ПРОЦЕССАХ СВАРКИ, НАПЛАВКИ, НАПЫЛЕНИЯ И РЕЗКИ МЕТАЛЛОВ, А ТАКЖЕ НАИБОЛЕЕ ХАРАКТЕРНЫЕ И ОПАСНЫЕ ВРЕДНЫЕ ВЕЩЕСТВА (III III), ПО КОТОРЫМ НЕОБХОДИМО ПРОВОДИТЬ КОНТРОЛЬ ВОЗДУШНОЙ СРЕДЫ В ЗОНЕ ДЫХАНИЯ РАБОТАЮЩИХ И В ВОЗДУХЕ РАБОЧЕЙ ЗОНЫ
| Технологический процесс | Вид сварочных (наплавочных) материалов, их тип и марка | Твердая фаза (ТССА) | Газы (ГССА) | ||||||||||||||||||||||
| Mn | F-ион фторидов | CrO3 | Cr2O3 | Ni | Cu | Al2O3 | MgO | Mo | V | V2O5 | Co | B2O3 | W | Zr | FeO (Fe2O3) | Si | TiO2 | ZnO | HF | CO | NO2 | O3 | |||
| хорошо раств. | плохо раств. | ||||||||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| А. Ручная дуговая сварка: | Электроды с покрытием (тип): | Во всех случаях сварки сталей, покрытых антикоррозийными цинкосодержащими грунтами, и оцинкованной стали | |||||||||||||||||||||||
| 1) Углеродистых, низколегированных и легированных конструкционных сталей, в том числе: | |||||||||||||||||||||||||
| а) углеродистых и низколегированных сталей общего назначения | 1) рутиловый (АНО-1, РБУ-4, АНО-4, АНО-13, АНО-18, АНО-19, АНО-20, ОЗС-4, ОЗС-6, ОЗС-12, ОЗС-17Н, МР-3, РБУ-5, НЭ-46 и др.) | III III III | + | + | + | III III III | III III III | + | |||||||||||||||||
| 2) ильменитовый (АНО-6, АНО-6М, АНО-17, ОСЗ-21, ОСЗ-23 и др.) | III III III | + | + | III III III | III III III | + | |||||||||||||||||||
| 3) кислый (СМ-5 и др.) | III III III | + | + | III III III | III III III | + | |||||||||||||||||||
| 4) кисло-целлюлозный (ОМА-2 и др.) | III III III | + | + | III III III | III III III | + | |||||||||||||||||||
| 5) целлюлозный (ВСЦ-4, ВСЦ-4А и др.) | III III III | + | + | III III III | III III III | + | |||||||||||||||||||
| 6) специальный (ОЗС-16 и др.) | III III III | + | + | III III III | III III III | + | |||||||||||||||||||
| 7) основной (УОНИ-13/45, УОНИ-13/55, УОНИ-13/55К, УОНИ-13/65, АНО-9, АНО-10, АНО-11, АНО-Д, АНО-Т, СМ-11, ДСК-50, УП-1/55, ОЗС-25, СК-2-50, ТМУ-21, ЭТМ-2У, ВСФ-65У, ВСФ-85, К-5А, ИТС-4 и др.) | III III III | III III III | III III III | + | + | III III III | III III III | + | |||||||||||||||||
 рутилово-основной (ОЗС-22Н и др.) рутилово-основной (ОЗС-22Н и др.) |
III III III | III III III | III III III | + | + | + | III III III | III III III | + | ||||||||||||||||
| б) сталей типа 09Г2С и 10ХСНД | основной (ОЗС-22Р) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | |||||||||||||
| (ОЗС-20Н, ОЗС-20Р) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||||
| (ВПА) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| в) низколегированной атмосферокоррозионностойкой стали типа 10ХНДП | основной (ОЗС-18) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||
| г) низколегированных перлитных сталей типа 06Г2 НАБ, 10ХГНМАЮ и др. | основной (ОЗС-24) | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| д) легированных сталей и сталей повышенной прочности | основной (УОНИ-13/85У, УОНИ-13/85) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| -«- (ВСФ-65, ВСФ-75) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| -«- (НИАТ-3М, ОЗШ-1) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| е) литейных сталей 27ХГСНМЛ, 35ХГСЛ, высокопрочных сталей 30ХГСА и др. | основной (Н-17/СВ-ВЛ-1ДГ) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | ||||||||||||
| ж) легированных теплоустойчивых сталей | 1) рутилово-основной (ОЗС-11) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | + | + | + | ||||||||||||
| 2) основной (ТЛМ-14, ЦУ-2ХМ, ЦЛ-38, УОНИ-13/45МХ) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | |||||||||||||
| -«- (ТМЛ-3У, ЦЛ-20, ЦЛ-39, ЦЛ-17, ЦЛ-20Б, ЦЛ-17-63) | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||
| -«- (ЦЛ-41) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| -«- (ЗИО-20) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| -«- (ЦЛ-20-67) | III III III | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||
| -«- (ЦЛ-21) | III III III | III III III | III III III | III III III | III III III | + | + | + | + | III III III | III III III | + | + | ||||||||||||
| -«- (ПТ-30) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||||
| -«- (РТ-45) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | III III III | III III III | + | + | |||||||||||
| з) высоколегированных сталей | 1) рутиловый (ОЗЛ-22, ОЗЛ-14А, ОЗЛ-17У) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | ||||||||||||||
| -«- (ОЗЛ-20) | III III III | III III III | III III III | III III III | + | + | + | + | III III III | III III III | + | ||||||||||||||
| 2) рутилово-основной (АНВ-34, ОЗЛ-36, ОЗЛ-9А, ОЗЛ-19, АНВ-13, АНВ-27, АНВ-23) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| -«- (АНВ-17, НИАТ-1, АНВ-26, ОЗЛ-27, ИМЕТ-10, ОЗЛ-28) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | III III III | III III III | + | + | |||||||||||
| -«- (ОЗЛ-17У, ОЗЛ-37-1) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | + | ||||||||||||||
| -«- (АНВ-28) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||
| -«- (АНВ-20) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | + | III III III | III III III | III III III | + | ||||||||||
| 3) основной (УОНИ-13/НЖ, ЛМЗ-1, УОНИ-13/НЖ2, ОЗЛ-5, ОЗЛ-7, ЦЛ-11, НБ-38, Л-40М, ЦТ-15, ЦТ-15-1, ОЗЛ-3, ЗИО-3, ЦЛ-9, ОЗЛ-6, ОЗЛ-8, ЗИО-8, ЦЛ-25, ГС-1, ЦЛ-33, ОЗЛ-29, НИИ-48Г, ОЗЛ-19, ЦТ-16, ЦТ-16-1, КТИ-7А, ОЗЛ-31) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| основной (НИАТ-6АМ, АНВ-26, НЖ-13, ОЗЛ-25Б, ЦТ-28, ОЗЛ-2, ОЗЛ-25, ОЗЛ-35, АНЖР-1, АНЖР-2, АНЖР-3У, НИАТ-5) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | + | III III III | III III III | + | + | ||||||||||
| -«- (ЗИ-ИМ-1) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | |||||||||||||
| -«- (КТИ-10, ЦЛ-32, КТИ-5) | III III III | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | + | III III III | III III III | + | + | |||||||||
| 4) специальный (ОЗЛ-23) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||||
| -«- (ОЗЛ-21) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | + | III III III | III III III | + | + | |||||||||||
| 2) Чугуна и заварка дефектов чугунного литья | Электроды с покрытием (тип): | ||||||||||||||||||||||||
| 1) кислый (ОЗЧ-2) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||||
| 2) основной (МНЧ-2, ОЗЧ-3, ОЗЧ-4) | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| -«- (ОЗЖЧ-1) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||||
| -«- (ЦЧ-4, ЦЧ-4А) | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| 3) Алюминия, заварка литья из алюминиево-кремнистых сплавов типа АЛ-4, АЛ-9 и др. | Электроды с покрытием (тип) специальный (ОЗА-1, ОЗА-2, 4ф-4аКр, А1-1Ф1) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | |||||||||||||
| 4) Алюминиево-марганцовистых бронз типа Бр АМц-9-2, алюминиево-никелевых бронз типа БрАЧ и др. и заварки дефектов литья | галогенидный (ЛКЗ-АБ) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| 5) Меди | специальный (Комсомолец-100) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| 6) Оловянисто-фосфористой бронзы и заварки дефектов литья | основной (ОЗБ-1) | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| 7) Латуни | основной (ОБ-5) | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | ||||||||||||||
| Никеля, биметалла (никель — сталь), сплава НМЖМЦ 28-2, 5-1 (монель) |
рутилово-основной (В-56У, «Прогресс-50», Н-50, Н-37, Имет-7, Имет-10) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| Б. Ручная дуговая наплавка углеродистых, низко-, средне- и высоколегированных сталей различного значения (в т.ч. штампов, режущего инструмента, жаропрочных деталей и пр.) | 1) Электроды с покрытием (тип): | ||||||||||||||||||||||||
| — основной (ОЗН-250У, ОЗН-300У, ОЗН-350У, ОЗН-400У, У340п/б) | III III III | III III III | III III III | + | + | III III III | III III III | + | |||||||||||||||||
| -«-«- (ОЗШ-3, 12АН/ЛИВТ) | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| -«-«- (С-1 (ГН-1)) | III III III | III III III | III III III | III III III | |||||||||||||||||||||
| -«-«- (УОНИ-13/НЖ, ОМГ-Н, ЗИО-8, ЦНИИН-4, ЦН-6Л, ЦН-16) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||||||
| -«- (ВСН-10, ЦН-12М, ЭА-898/21Б, ЭА-855/51, УОНИ-13/Н1-БК, ЭА-582/23, ЭА-400/10У, ЭА-400/10Т, ЭА-395/9 | III III III | III III III | III III III | III III III | III III III | III III III | + | + | Nb+ | III III III | III III III | + | + | ||||||||||||
| -«»-«»- (ЭН-60М, НР-70, 13КН/ЛИВТ, ОЗШ-1) | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||||
| -«»-«»- (ОЗИ-3, ОЗШ-2, ТК-3Н) | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||
| -«»-«»- (УОНИ-13/4Х10В5МФ, ВПИ-1) | III III III | III III III | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||
| -«»-«»- (ОЗШ-4) | III III III | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | III III III | + | + | III III III | III III III | + | + | |||||||||
| -«»-«»- (ЦИ-1М, ИТ-10, ВСН-6) | III III III | III III III | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | |||||||||||||
| -«»-«»- (ЖСН-60Р) | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||
| -«»-«»- (ОЗИ-4, ОЗИ-5) | III III III | III III III | III III III | III III III | III III III | + | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | |||||||||
| -«»-«»- (ЦН-2) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| -«»-«»- (ЭНУ-2, ВСН-8, Т-590, Т-620) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | + | + | ||||||||||||
| -«»-«»- (ВСН-9) | III III III | III III III | III III III | III III III | III III III | III III III | III III III | + | + | III III III | III III III | + | + | ||||||||||||
| 2) Литые прутки для дуговой наплавки сталей неплавящимся (вольфрамовым электродом) | |||||||||||||||||||||||||
| — (ПрС2, ПрС1) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | ||||||||||||||||
| — (ПрС27) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | ||||||||||||||||
| — (Пр-В3К) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | ||||||||||||||||
| — (Пр-В3К-Р) | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | |||||||||||||||
| 3) Смеси порошков для наплавки сталей неплавящимся электродом | |||||||||||||||||||||||||
| — (С-2М) | III III III | III III III | III III III | + | + | + | III III III | III III III | |||||||||||||||||
| — (БХ, КБХ) | III III III | III III III | III III III | + | + | + | III III III | III III III | |||||||||||||||||
| — (ФБХ6-2) | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | ||||||||||||||||
| — (ПС-15-30, ПС-14-60, ПС-14-80) | III III III | III III III | III III III | III III III | III III III | III III III | + | + | + | III III III | III III III | ||||||||||||||
| Вольфрамовые электроды: | |||||||||||||||||||||||||
| РУЧНАЯ ДУГОВАЯ СВАРКА И НАПЛАВКА МЕТАЛЛОВ НЕПЛАВЯЩИМСЯ (ВОЛЬФРАМОВЫМ) ЭЛЕКТРОДОМ В ЗАЩИТНЫХ ГАЗАХ (АРГОН, ГЕЛИЙ, АЗОТ), ГАЗОВЫХ СМЕСЯХ. | — ЭВИ-1, ЭВИ-2 | III III | +V2O3 | III III | III III | ||||||||||||||||||||
| — ЭВЛ | III III | +La | III III | III III | |||||||||||||||||||||
| — ЭВЧ | III III | III III | III III | ||||||||||||||||||||||
| — ЭВТ | III III | +Th | III III | III III | |||||||||||||||||||||
| — Релит-3, Релит-Т3, АНЛ-3 и др. | III III | III III | III III | ||||||||||||||||||||||
| В том числе: | |||||||||||||||||||||||||
| 1. Сталей различного назначения <**> | Присадочные проволоки, содержащие легирующие элементы, обозначаемые в наименовании буквами <*>: | ||||||||||||||||||||||||
| Б — ниобий | + | + | III III | III III | |||||||||||||||||||||
| Г — марганец | III III | + | + | III III | III III | ||||||||||||||||||||
| В — вольфрам | + | III III | III III | III III | |||||||||||||||||||||
| Д — медь | III III | + | + | III III | III III | ||||||||||||||||||||
| К — кобальт | III III | + | + | III III | III III | ||||||||||||||||||||
| М — молибден | III III | + | + | III III | III III | ||||||||||||||||||||
| Н — никель | III III | + | + | III III | III III | ||||||||||||||||||||
| Р — бор | III III | + | + | III III | III III | ||||||||||||||||||||
| С — кремний | + | + | III III | III III | III III | ||||||||||||||||||||
| Т — титан | + | III III | + | III III | III III | ||||||||||||||||||||
| Ф — ванадий | III III | III III | + | + | III III | III III | |||||||||||||||||||
| Х — хром | III III | III III | + | + | III III | III III | |||||||||||||||||||
| Ю — алюминий | III III | + | + | III III | III III | ||||||||||||||||||||
| Ц — цирконий | + | III III | + | III III | III III | ||||||||||||||||||||
| 2. Алюминия и его сплавов в среде аргона, гелия или их смесей | Присадочные электродные проволоки: | ||||||||||||||||||||||||
| В том числе, марок: | |||||||||||||||||||||||||
| а). АД0 | а). СвАД0 | III III | + | III III | III III | ||||||||||||||||||||
| б). АД1 | б). СвАК5 | III III | III III | + | III III | III III | |||||||||||||||||||
| — СвАМг3 | III III | III III | + | + | III III | III III | |||||||||||||||||||
| — СвАМг6 | + | III III | III III | + | III III | III III | |||||||||||||||||||
| — СвАМц | + | III III | + | III III | III III | ||||||||||||||||||||
| в). АМц | в). СвАК5, | + | III III | III III | + | III III | III III | ||||||||||||||||||
| СвАМг3, СвАМг6 и СвАМц то же, что и в п. «б» | + | III III | III III | + | + | III III | III III | ||||||||||||||||||
| г). АМг3 | г). СвАК5, | + | III III | III III | III III | + | + | III III | III III | ||||||||||||||||
| СвАМг3, | + | III III | III III | + | + | III III | III III | ||||||||||||||||||
| СвАМц | + | III III | III III | + | + | III III | III III | ||||||||||||||||||
| д). АМг6 | д). СвАМг6 | + | III III | III III | + | +Be? | III III | III III | |||||||||||||||||
| е). АВ | е). СвАК5 | + | + | III III | III III | III III | + | + | III III | III III | |||||||||||||||
| ж). АД 33 | ж). СвАК5 | + | III III | III III | III III | + | + | III III | III III | ||||||||||||||||
| 3. Магния и его сплавов, в том числе: | Присадочные электродные проволоки: | ||||||||||||||||||||||||
| МА2-1, МА-8, МА-9, МА-11 | — МА2-1, МА-5, МА-9 | III III | + | III III | III III | ||||||||||||||||||||
| — МЛ-7 | + | + | III III | + | + | + | III III | III III | |||||||||||||||||
| 4. Меди и бронз | Присадочные электродные проволоки: | ||||||||||||||||||||||||
| — раскисленная медь | III III | + | III III | III III | |||||||||||||||||||||
| — медно-никелевая МНЖКТ 5-1-0,2-0,2 | + | III III | III III | + | + | + | III III | III III | |||||||||||||||||
| — бронзовая Бр.КМц 3-1, Бр.ОЦ4-3 | + | III III | + | + | III III | III III | |||||||||||||||||||
| 5. Титана и сплавов на его основе | Присадочные электродные проволоки: | ||||||||||||||||||||||||
| — ВТ1-00 | + | III III | III III | III III | |||||||||||||||||||||
| — ВТ2 | + | + | III III | III III | III III | ||||||||||||||||||||
| — СПТ-2 | + | + | + | + | III III | III III | III III | ||||||||||||||||||
| 6. Никеля и его сплавов, в том числе: | Присадочные электродные проволоки: | ||||||||||||||||||||||||
| а). ЭП 495, ЭП 490 | а). ЭИ639, Н70М27 | III III | + | + | III III | III III | |||||||||||||||||||
| б). ОХ15Н55М16В | б). ЭП 567 | + | + | III III | + | + | III III | III III | |||||||||||||||||
| Г. МЕХАНИЗИРОВАННАЯ (ПОЛУАВТОМАТИЧЕСКАЯ И АВТОМАТИЧЕСКАЯ) ДУГОВАЯ СВАРКА И НАПЛАВКА: | |||||||||||||||||||||||||
| 1. Сталей различного назначения в защитной среде CO2 и газовых смесях | Проволоки сплошного сечения, в том числе: | ||||||||||||||||||||||||
| 1.1. углеродистых и низколегированных сталей | 1.1. Св-12ГС, Св-08Г2С, Св-08ГС | III III | + | + | III III | + | + | ||||||||||||||||||
| 1.2. низколегированных сталей 10ХСНД | 1.2. Св-18ХГС, | III III | + | + | + | + | III III | + | + | ||||||||||||||||
| Св-08ХГ2С | III III | + | + | + | III III | + | + | ||||||||||||||||||
| 1.3. низколегированных теплоустойчивых сталей типа 30ХГСА, МА, 20ХМА, МФЛ и др. | 1.3. Св-08ХГСМА, Св-10ХГ2СМА, Св-08ХГСМФА, Св-08Х3Г2СМ | III III | + | + | + | III III | + | + | |||||||||||||||||
| 1.4. средне- и высоколегированных сталей | 1.4. Св-08Х20Н9Г7Т, Св-08Х21Н10Г6 | III III | + | III III | + | + | III III | + | + | ||||||||||||||||
| — Св-20Х13 | III III | III III | III III | + | + | III III | + | + | |||||||||||||||||
| — Св-08Х16Н8М2, Св-06Х24Н6ТАФМ | + | III III | III III | III III | + | + | + | III III | + | + | |||||||||||||||
| — Св-01Х19Н9, Св-Х18Н8Г2Б, Св-07Х18Н9ТЮ, Св-06Х19Н9Т, Св-08Х19Н9Ф2С2 и др. | + | III III | III III | III III | + | + | III III | + | + | ||||||||||||||||
| 1.5. сварка углеродистых и низколегированных сталей | 2. Порошковые проволоки самозащитные: | ||||||||||||||||||||||||
| — рутил-органические (ПП-АН1, ПП-1ДСК) | III III | + | + | + | III III | + | |||||||||||||||||||
| — карбонатно-флюоритные (ПП-АН3, ПП-АН7, ПП-АН2М, СП-1, СП-2, ППВ-5, ПП-АН-11, ПП-АН23 | III III | III III | III III | + | + | + | + | III III | + | + | |||||||||||||||
| — флюоритный тип (ПП-2ДСК) | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||||
| 2.1. Порошковые проволоки для сварки в защищенной среде CO2 (тип): | |||||||||||||||||||||||||
| — рутиловый (ПП-АН8, ПП-АН10, ПП-АН13, ПП-АН21 и др.) | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||||
| — рутил-флюоритный (ПП-АН22, ПП-АН4, ПП-АН9, ПП-АН18, ПП-АН20 и др.) | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||||
| — карбонатно-флюоритный (ПП-АН5) | III III | III III | III III | + | + | + | III III | III III | + | ||||||||||||||||
| 2.3. Специального назначения самозащитные: | |||||||||||||||||||||||||
| — карбонатно-флюоритные (ПП-АН15, ПП-АН19, ПП-АН19Н, ПП-АНЗС, ПП-АН6) | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||||
| — тот же тип (ПП-АНВ1, ПП-АНВ2, ПП-АНА1) | III III | III III | III III | III III | III III | III III | + | + | +Nb | III III | + | + | |||||||||||||
| — (ПП-АНВ3) | III III | III III | III III | III III | III III | + | + | III III | + | + | |||||||||||||||
| 1.6. Наплавка поверхностных слоев на детали | 4. Порошковая проволока наплавочная самозащитная: | ||||||||||||||||||||||||
| — ПП-Нп-10Х14Т | III III | III III | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||
| — ПП-Нп-30Х5Г2СМ | III III | III III | III III | III III | III III | + | + | + | + | III III | + | + | |||||||||||||
| — ПП-Нп-200Х17ТС2ГР | III III | III III | III III | III III | III III | III III | + | + | + | III III | + | + | |||||||||||||
| — ПП-Нп-25ХБФМС | III III | III III | III III | III III | III III | + | III III | III III | + | + | III III | + | + | ||||||||||||
| — ПП-Нп-250Х10Б8СТ | III III | III III | III III | III III | +Nb | III III | + | + | |||||||||||||||||
| — ПП-Нп-80Х20РЗТ | III III | III III | III III | III III | III III | III III | + | + | + | III III | + | + | |||||||||||||
| — ПП-Нп-10Х15Н2Т | III III | III III | III III | III III | III III | III III | + | + | + | III III | + | + | |||||||||||||
| — ПП-Нп-14СТ | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||||
| — ПП-Нп-19СТ, ПП-Нп-50Х3СТ | III III | III III | III III | III III | III III | + | + | + | III III | + | + | ||||||||||||||
| 1.7. Сварка (наплавка) и заварка дефектов чугунного литья | а) Проволока сплошного сечения ПАНЧ-11 | + | III III | + | + | + | + | + | + | ||||||||||||||||
| б) Порошковые проволоки самозащитные: (ПП-АНЧ2, ПП-АНЧ5) | III III | III III | III III | + | + | + | + | III III | + | + | |||||||||||||||
| 1.8. Сварка и наплавка сталей под флюсом | а) флюсы наплавленные (АН-60, АН-348А, АН-348В, ОСЦ-45, АН-47, АН-22, АН-65, АН-17М, АН-43, АН-26С, АН-20С, АН-8, АН-25 и др.) | III III | III III | III III | + | + | + | + | + | III III | + | ||||||||||||||
| б) флюсы керамические (АНК-30) | III III | III III | III III | + | + | + | III III | + | |||||||||||||||||
| (АНК-35 и др.) | III III | III III | III III | + | + | III III | + | ||||||||||||||||||
| 1.9. Сварка цветных металлов и сплавов под флюсом, в том числе: | Флюсы плавленные и керамические: | ||||||||||||||||||||||||
| а) меди и латуни | а) АН-26С, АН-60, КМ-1, К-13 | III III | III III | III III | + | + | + | III III | + | ||||||||||||||||
| б) никеля | б) ЖМ-1 | III III | III III | III III | + | + | III III | + | |||||||||||||||||
| в) алюминия | в) ЖА-64, ЖА-64А, МАТИ-10 | + | III III | III III | + | III III | |||||||||||||||||||
| 8. Электрошлаковая сварка меди | Флюс АН-М10 | III III | III III | III III | + | + | III III | + | |||||||||||||||||
| 9. Полуавтоматическая и автоматическая сварка и наплавка цветных металлов (алюминия, меди, титана и др.) в защитных газах | См. раздел «В» п. п. 2, 3, 4, 5 и 6 | ||||||||||||||||||||||||
| Д. ПЛАЗМЕННАЯ СВАРКА И РЕЗКА МЕТАЛЛОВ | Выбор ингредиентов ТССА при контроле воздушной среды при плазменной сварке осуществляется с учетом состава используемого электродного (присадочного) материала, а при резке — исходя из состава разрезаемого металла <*> | III III | III III | ||||||||||||||||||||||
| Е. ГАЗОВАЯ (АЦЕТИЛЕН-КИСЛОРОДНАЯ) РЕЗКА И СВАРКА МЕТАЛЛОВ | Выбор ингредиентов ТССА тот же, что и при плазменной сварке и резке металлов | III III | III III |
<*> Содержание легирующих элементов, если оно не превышает 1%, ставится после соответствующей буквы в целых единицах (проценты). Первые две цифры (для конструкционных марок сталей) обозначают число сотых процентов углерода. Например, марка проволоки (или стали) 12ХНЗ означает, что в стали содержится 0,12% C, около 1% Cr и около 3% никеля.
<**> В случае одновременного присутствия в проволоке нескольких легирующих элементов аналитическому определению в воздухе подлежат те из них, которые относятся к I — II классам опасности. Железо и вольфрам присутствуют во всех образующихся ТССА.
<*> Дополнительные сведения о составе ТССА содержатся в «Санитарных правилах на устройство и эксплуатацию оборудования для плазменной обработки материалов» N 4053-85 Минздрава СССР.
Приложение 7
ПРИНЦИПИАЛЬНАЯ СХЕМА ГАЗОХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ ДЛЯ ОПРЕДЕЛЕНИЯ ОКСИДА УГЛЕРОДА (II)

1 — кран-дозатор
2 — хроматографическая колонка
3 — метанатор
4 — детектор
5 — усилитель
6 — потенциометр
Руководство по контролю вредных веществ в воздухе рабочей зоны
МОСКВА« ХИМИЯ» 1991
дины и конца процесса, а также с учетом продолжительности выделения наибольшего количества токсичных веществ.
Для получения достоверных результатов при санитарнохимических исследованиях воздушной среды в любой точке на каждой стадии технологического процесса или отдельной операции должно быть последовательно отобрано не менее пяти проб воздуха.
Вычисляют среднее арифметическое значение (концентрация С, ыг/м6) и доверительный интервал (е, %):
С= (С|-(-Сг+Сз+Сч+Сб)/5,
е
(^макс ^мнн) * 60/С,
где С|,…,Сб — концентрация в отдельных пробах; Смакс— максимальная концентрация в отобранных пробах; Смнн— минимальная концентрация в отобранных пробах.
Если полученное значение доверительного интервала равно или меньше 40%, то значение средней арифметической считается достоверным. Если вычисленный доверительный интервал превышает 40%, должны быть отобраны дополнительные пробы, число которых п определяют по формуле:
я=[5.8(Снвкс-Синн)/С]-5.
Выбор мест отбора проб воздуха. В новых или ранее неизученных в гигиеническом плане производствах, воздушная среда на которых может загрязняться токсическими веществами, санитарный контроль следует проводить преимущественно на всех рабочих местах с постоянным и временным пребыванием работающих.
На основе данных по исследованию загрязненности воздуха рабочей зоны в комплексе с данными по оценке технологического процесса, оборудования, вентиляционных устройств определяют наиболее неблагоприятные в санитарно-гигиеническом отношении рабочие места, на которых в дальнейшем отбирают пробы воздуха.
Санитарный контроль загрязнений воздушной среды осуществляют выборочно на отдельных рабочих местах, стадиях или операциях, если на обследуемом участке, характеризующемся постоянством технологического процесса, достаточно идентичное оборудование или одинаковые рабочие места, на которых выполняют одни и те же операции. При этом отбор проб следует проводить на рабочих местах, расположенных в центре и по периферии помещения или на открытой площадке с оборудованием. При выборе точек отбора проб основное внимание следует уделять рабочим местам по основным профессиям.
Пробы отбирают с учетом технологических операций, при которых возможно наибольшее выделение в воздух рабочей зоны вредных веществ, например: у аппаратуры и агрегатов в
10
период наиболее активных химических и термических процессов (электрохимических, пиролитических и др.); на участках загрузки и выгрузки веществ, затаривания продукции; на участках транспортировки, размола и сутки сыпучих, пылящих материалов; в местах наиболее вероятных источников выделения при перекачке жидкостей и газов (насосные, компрессорные) и др.; в местах отбора технологических проб, необходимых для анализа; на участках, плохо вентилируемых, необходимо проводить санитарно-химический анализ воздуха рабочей зоны на основных местах пребывания работающих в период проведения планового ремонта технологического, санитарно-технического и другого оборудования, если эти операции могут сопровождаться выделением вредных веществ, в период реконструкции, если при этом часть оборудования продолжает эксплуатироваться.
Периодичность отбора проб воздуха для каждого вещества в каждой точке устанавливают в зависимости от характера технологического процесса (непрерывного, периодического), класса опасности и характера биологического действия производственной среды, уровня загрязнения, времени пребывания обслуживающего персонала на рабочем месте.
При возможном поступлении в воздух рабочей зоны производственных помещений вредных веществ с остронаправленным механизмом действия пробы следует отбирать с применением систем автоматических приборов. При отсутствии приборов непрерывного контроля как временная мера допускается при согласовании с органами санитарного надзора периодический отбор проб воздуха для определения вещества с остронаправленным механизмом действия. Для остальных веществ периодичность контроля следует устанавливать в зависимости от класса опасности вредного вещества: для веществ I класса опасности — не реже одного раза в 10 дней; для веществ II класса — не реже одного раза в месяц; для веществ III и IV классов — не реже одного раза в квартал.
В зависимости от конкретных условий производства периодичность контроля может быть изменена по согласованию с санитарно-эпидемиологической службой.
Измерение среднесменных концентраций предусмотрено для веществ, которые имеют соответствующий норматив — ПДК с.с. Для характеристики уровня среднесменных концентраций, воздействующих на рабочих одной профессиональной группы, необходимо провести обследование не менее 5 человеко-смен. Среднесменную концентрацию в зоне дыхания работающих измеряют приборами индивидуального контроля при непрерывном или последовательном отборе проб воздуха в течение всей смены или не менее 75% ее продолжительности.
Продолжительность отбора одной пробы и число проб за смену зависят от методики и концентрации токсического вещества
11
в воздухе. В некоторых случаях среднесменную концентрацию Ссс (мг/м*) вычисляют по результатам разовых измерений на отдельных местах пребывания рабочих с учетом хрономегражных данных и рассчитывают по формуле:
^СС~ (Clfl+^2 *2 + —+ СП/Л)/ ( Л+/2+.,
где Ci, Сг,…,Сл среднеарифметические значения разовых измерений концентраций вредных веществ на отдельных стадиях технологического процесса, мг/м4; — продолжительность отдельных стадий технологического процесса, мин.
Среднеарифметическое значение из результатов разовых измерений следует вычислять с учетом нулевых значений измерений.
В настоящее время для измерения среднесменных концентраций химических веществ разработано новое устройство — пассивный дозиметр.
Выбор способа отбора обычно определяется природой анализируемых веществ, наличием сопутствующих примесей и другими факторами. Для обоснованного выбора способа отбора проб необходимо иметь четкое представление о возможных формах нахождения токсических примесей в воздухе. Микропримеси вредных веществ в воздухе могут находиться в виде газов (аммиак, дивинил, озон и др.), в виде паров — преимущественно вещества, представляющие собой жидкость с температурой кипения до 230—250°С (ароматические хлорированные и алифатические углеводороды, низшие ациклические спирты, кислоты и др.), а также некоторые твердые вещества, обладающие высокой летучестью (иод, нафталин, фенол). Иногда вещества могут находиться в воздухе одновременно в виде паров и аэрозолей. Это преимущественно жидкости с высокой температурой кипения (дибутилфталат, диметилтерефталат, капролактам и др.). Попадая в воздух, их пары конденсируются с образованием аэрозоля конденсации.
Аэрозоли конденсации образуются также при некоторых химических реакциях, приводящих к появлению новых жидких или твердых фаз. Например, при взаимодействии триоксида серы (серного ангидрида) с влагой образуется туман серной кислоты; тетрахлорид титана с влагой воздуха образует дым диоксида титана; аммиак и хлороводород образуют дым хлорида аммония. Конденсационное происхождение имеют также аэрозоли, образующиеся при сварочных работах и других высокотемпературных процессах, сопровождающихся расплавлением и испарением металлов. Например, свинец, поступающий в воздушную среду в виде паров при нагреве свинца и его сплавов до температуры выше 400°С, в воздухе рабочей зоны находится в виде аэрозоля конденсации.
Наряду с аэрозолями конденсации в различных производственных процессах (например, при механическом измельчении твердых веществ и распылении жидкостей) образуются аэрозоли дезинтеграции с более грубой дисперсностью. Причем при 12
значительной летучести дисперсной фазы аэрозоля возможно одновременное присутствие аэрозоля и паров (пульверизацион-ная окраска изделий).
Правильное установление агрегатного состояния вредного вещества в воздухе способствует правильному выбору фильтров и сорбентов и уменьшению погрешности определения, связанной с пробоотбором. Для предварительной оценки агрегатного состояния примесей в воздухе необходимо располагать сведениями об их летучести — максимальной концентрации паров, выраженной в единицах массы на объем воздуха при данной температуре (см. Приложение 1).
Летучесть L (мг/л) рассчитывают по формуле:
L =16PAf/(273+0» (1.1)
где Р — давление насыщенного пара при данной температуре, мм рт. ст.; М — молекулярная масса вещества; t — температура, °С.
В справочной литературе не всегда имеются данные о давлении паров, поэтому предложена формула расчета ориентировочного давления насыщенных паров при различных температурах [3]:
]g/>=2,763—0,019/кип+0,024*. (1.2)
Формула выражает корреляционную связь между температурой кипения вещества /кип, давлением насыщенного пара Р, мм рт. ст. при / °С и температурой внешней среды t°С.
Таким образом, зная температуру кипения вещества, можно рассчитать давление насыщенного пара при различных температурах и затем летучесть вещества с погрешностью до ±30—40%.
При отсутствии данных о давлении насыщенных паров исследуемого вещества летучесть последнего можно также ориентировочно установить методом химического анализа. Для этого в газометр вместимостью 5—20 л помещают избыток вещества и, периодически отбирая пробы аспирационным или вакуумным способом, определяют концентрацию продукта в воздухе при заданной температуре рекомендуемым методом.
При классификации вредных веществ по их агрегатным состояниям в воздухе необходимо учитывать помимо летучести их санитарные нормы (ПДК), например ртуть (/КИП357°С) по сравнению с бутилацетатом (*кип 126°С) можно считать малолетучей жидкостью; летучести этих веществ при 20°С соответственно равны 15 и 20 000 мг/ма. Однако в связи с большой разницей в ПДК (0,01 и 200 мг/м3 соответственно) максимальное содержание в воздухе малолетучей ртути при 20°С может превышать санитарную норму в 1500 раз, а содержание паров бутилового спирта только в 250 раз. Поэтому агрегатное состояние рекомендуется оценивать по отношению летучести вещества при 20°С (mt/mj) к его ПДК—/,2о/ПДК [4,5]. Если относительная летучесть вещества (например, серной кислоты, дино-
13
нилфталата) при 20°С ниже ПДК (в 10 и более раз), то наличием паров можно пренебречь. В этом случае определяют лишь содержание в воздухе аэрозоля. При значительном превышении ПДК (в 50 и более раз) определяют только пары (например, этилмеркурхлорид, нафталин и др.).
К парам и аэрозолям следует относить вещества, летучесть которых при 20°С составляет от 10 до 50 ПДК (50>£/ПДК>10). По данным зарубежных исследователей, при отношении равновесной концентрации пара соединения при 25°С к ПДК от 0,05 до 50 следует учитывать наличие паров и аэрозольных частиц. При отношении меньше 0,05 следует учитывать лишь наличие паров вещества.
При проведении санитарно-химических исследований на производстве пробы воздуха отбирают преимущественно аспирационным способом путем пропускания исследуемого воздуха через поглотительную систему (жидкая поглотительная среда, твердые сорбенты или фильтрующие материалы). Минимальная концентрация вещества, поддающаяся четкому и надежному определению, зависит от количества отбираемого воздуха. Аспирация излишних объемов воздуха приводит к неоправданным потерям рабочего времени, при недостаточном объеме воздуха снижается точность анализа, а иногда вообще оказывается невозможным проведение количественных определений.
В сборниках Методических указаний по измерению концентраций вредных веществ в воздухе указаны объемы воздуха, необходимые для определения 1/2 ПДК. Однако при проектировании вентиляционных систем производственных помещений и особенно при выполнении работ по наладке вентиляционных установок имеется необходимость в определении значительно меньших концентраций вредных веществ. В то же время в некоторых случаях (например, при контроле промышленных выбросов) вредные вещества могут находиться в концентрациях, значительно превышающих предельно допустимые. Тогда следует брать меньшие пробы воздуха.
Оптимальный объем воздуха V, необходимый для определения токсической примеси с заданной точностью, можно рассчитать по следующей формуле:
V=aVJVnKC, (1.3)
где а — нижний предел обнаружения в анализируемом объеме пробы, мкг; V0— общий объем пробы, мл; Vn—объем пробы, взятой для анализа, мл; С — предельно допустимая концентрация, мг/м3; К — коэффициент, соответствующий долям ПДК (1/4, 1/2, 1 ПДК и т.д.).
Объемы воздуха, необходимые для анализа, пересчитывают по мере усовершенствования методов анализа, а также при изменении ПДК.
Аспирационный способ отбора проб применяют в основном при гигиенической оценке длительных стадий технологического
14
процесса. Время отбора для определения максимальных разовых концентраций ограничивается 15 мин [2]. Однако в настоящее время все большее внимание придают среднесменным концентрациям, позволяющим определить поглощенную дозу вредного вещества. Среднесменные пробы берут индивидуальными пробоотборниками с сорбционными трубками, закрепленными на воротнике спецодежды работающего. Результаты анализа при этом могут быть получены непосредственно на месте при использовании линейно-колористических методов с помощью индикаторных трубок или в лаборатории после извлечения поглощенных примесей из сорбента.
При кратковременных технологических процессах, если позволяет чувствительность методов анализа, используют быстрые способы отбора проб (в газовые пипетки, шприцы). При недостаточной чувствительности метода и кратковременных операциях прибегают к концентрированию веществ из воздуха с помощью поглотительной системы. При этом в некоторых случаях (при отборе на фильтр) допускается повышение скорости пробоот-бора. Пробу можно отбирать в одну и ту же поглотительную систему при повторении операции.
Многообразие вредных веществ и агрегатных состояний в воздухе обусловливает использование различных поглотительных систем, обеспечивающих эффективное поглощение микропримесей.
1.1. ОТБОР ПРОБ В ЖИДКИЕ СРЕДЫ
Отбор парогазовых веществ в жидкие поглотительные среды — наиболее распространенный способ. Анализируемые вещества растворяются или вступают в химическое взаимодействие с поглотительной средой (хемосорбция), которая обеспечивает полноту поглощения за счет образования нелетучих соединений. При этом упрощается подготовка пробы к анализу, который обычно проводят в жидкой фазе. Кроме того, правильный выбор соответствующего растворителя дает возможность провести раздельное определение веществ в процессе отбора проб.
Отбор проб в растворы осуществляют аспирацией исследуемого воздуха через поглотительный сосуд (абсорбер) с каким-либо растворителем (органические растворители, кислоты, спирты, вода и др.). Скорость пропускания воздуха может меняться в широких пределах — от 0,1 до 100 л/мин.
Полнота поглощения зависит от многих факторов, в том числе от конструкции поглотительных сосудов. На рис. 1.1 —1.4 представлены абсорберы, широко используемые в практике санитарного контроля. Наибольшее распространение получили абсорберы со стеклянными пористыми пластинками, поглотительные сосуды Рыхтера, Зайцева. Теоретические основы абсорбции газов описаны в монографии [6].
15
|
Рис. 1.1. Поглотительный сосуд Зайцева Рис. 1.2. Поглотительные сосуды с пористой пластинкой |
Для физической абсорбции важно, чтобы поверхность соприкосновения фаз была наибольшей. В поглотителях с пористой пластинкой этот эффект достигается за счет уменьшения пузырьков воздуха при прохождении его через пористый фильтр, вследствие чего увеличивается контакт воздуха с раствором, а скорость аспирации воздуха может быть повышена до 3 л/мин.
Увеличение поверхности контакта может быть достигнуто также в результате увеличения длины пути прохождения пузырьков воздуха через раствор. Так, в поглотительных сосудах Зайцева высота столба растворителя составляет около 10 см. Однако предельная скорость просасывания воздуха через такой поглотитель не превышает 0,5—0,6 л/мин.
При отборе проб в поглотительные сосуды Рыхтера (пяти моделей), в которых используют эффект эжекции, скорость аспирации воздуха может достигать 100 л/мин.
Более эффективным является поглощение, основанное на химических реакциях исследуемых веществ с поглотительной жидкостью. Например, для поглощения аммиака и аминов применяют разбавленную серную кислоту, для поглощения фенола — раствор щелочи (гидрокарбонат натрия).
Для проверки эффективности работы поглотительного сосуда к нему присоединяют последовательно еще один или два поглотителя. Пробу воздуха с известным содержанием вредного вещества пропускают через все абсорберы и затем поглотительные растворы из каждого сосуда анализируют.
«Проскок» К (в %) вычисляют по формуле:
/С=Л2/(Л|+Л2). 100, (1.4)
где А2 — масса вещества во втором абсорбере, мкг; А — масса вещества в первом абсорбере, мкг.
(1.5)
Степень поглощения Э (в %) вычисляют по формуле:
3=100 — к.
16
|
Рис. 1.4. Поглотительные сосуды для зерненых сорбентов «в кипящем слое»: а— сосуд Яворовской; б— видоизмененный сосуд Зайцева |
Если в первом сосуде абсорбировалось около 95% исследуемого вещества, эффективность поглощения можно считать достаточной. В случае неполного поглощения вещества в первом поглотителе необходимо использовать второй поглотитель.
При применении в качестве поглотительной жидкости летучих органических растворителей отбор проб рекомендуется проводить в охлаждаемый абсорбер. В противном случае происходит испарение жидкости, и погрешность количественного анализа заметно возрастает.
1.2. ОТБОР ПРОБ НА ТВЕРДЫЕ СОРБЕНТЫ
Гранулированные сорбенты для отбора паров химических веществ из воздуха начали применять в конце 60-х годов в связи с широким развитием газовой хроматографии.
Способ отбора проб воздуха в жидкости для газохроматографического анализа в большинстве случаев неприемлем, так как не позволяет проводить концентрирование веществ из большого объема воздуха вследствие улетучивания растворителей и связанных с этим потерь анализируемых веществ. Кроме того, жидкие поглотительные среды для одновременного пробоотбора химических веществ с разными функциональными группами не обеспечивают эффективного поглощения для каждой труп ы веществ.
Применение твердых сорбентов дает возможность увеличить скорость пропускания воздуха (по сравнению с пропусканием через жидкость) и за короткое время накопить исследуемое
17
вещество в количестве, достаточном для его определения. Твердые сорбенты позволяют также осуществлять избирательную сорбцию одних веществ в присутствии других, кроме того, твердые сорбенты удобны как в работе, так и при транспортировке и хранении отобранных проб.
В качестве сорбентов применяют силикагели, молекулярные сита, активный уголь, пористые полимерные сорбенты и другие вещества, т.е. сорбенты с активной поверхностью. Кроме того, используют наполнители хроматографических колонок.
Твердые сорбенты, применяемые для отбора проб воздуха, должны обладать механической прочностью, иметь небольшое сродство к водяным парам (т.е. плохо сорбировать их), легко активироваться, иметь максимальную сорбционную способность по отношению к анализируемым веществам, а при анализе легко десорбировать поглощенное вещество, иметь однородную структуру поверхности. Сорбционно-десорбционные свойства сорбента определяются не только полярностью его поверхности, но и эффективным радиусом пор и их формой. Поры с эффективным радиусом менее 2 нм называют микропорами. Они играют для пробоотбора наиболее существенную роль, а их объем, выраженный в см3/г сорбента, определяет его сорбционную способность. У сорбентов с очень малой сорбционной способностью общий объем микропор составляет менее 0,1 см3/г, а самые эффективные сорбенты имеют общий объем пор 0,5—0,6 см3/г. Объем промежуточных пор сорбента с радиусом меньше 100 нм и макропор — меньше 200 нм не является значимым для сорбентов, предназначенных для пробоотбора. При очень низких концентрациях паров и газов химических веществ в воздухе рабочей зоны адсорбция осуществляется преимущественно адсорбционными центрами в микропорах.
Для анализа промышленного воздуха применяют три группы твердых адсорбентов, однако ни один из сорбентов не является универсальным. Первая группа представляет собой гидрофильные неорганические материалы типа силикагелей и молекулярных сит. Вторая группа — гидрофобные неорганические материалы — активные угли. К третьей группе относят синтетические макропористые органические материалы с высокой степенью гидрофобности и небольшой удельной поверхностью — это пористые полимеры, например хромосорбы, порапаки и др.
Силикагели. Силикагели (SiC>2- хНгО) представляют собой гидрофильные сорбенты с высокоразвитой капиллярной структурой геля. Содержание воды в нем около 5% (масс.). Вода является частью структуры геля и связана с атомами кремния в форме активных гидроксильных групп. Адсорбционная способность силикагеля обусловлена наличием на его поверхности силаноль-ных групп Si — ОН, способных к образованию водородных связей с молекулами сорбата. На адсорбционные свойства большое
18
влияние оказывает структура сорбента. Микропористые сорбенты имеют большую удельную поверхность — от 600 до 800 м2/г, макропористые — от 120 до 500 мг/г. Некоторые характеристики стандартных силикагелей приведены ниже:
Эффективный диаметр преобладающих пор, нм…….. 2,5 4 6 10 100 1000
Объем пор, см3/г…… 0,45 0,65 0,75 1,00 0,65 0,50
Удельная поверхность, м2/г . . . 750 650 500 400 25 2
В табл. 1.1 приведены основные типы силикагелей и их характеристики.
Силикагели предназначены для адсорбции из воздуха паров полярных органических соединений, веществ с гидроксильными группами и других кислородсодержащих соединений. Ненасыщенные соединения обычно сорбируются лучше, чем их насыщенные производные.
В работах Брокманна [7] и Стюарта [8] показана зависимость снижения адсорбционной способности у ряда заместителей (R — алифатический радикал, Аг — ароматический радикал):
Ряд Стюарта Ряд Стюарта
Ряд Брокманна Ряд Брокманна
Аг—ОН Аг—ОН NH—COR —NH2
R—СООГМН2 R—ГМ02 R—ОСНз R-H
R—СООН R—CONH2 R—ОН R—NH2
Микропористые силикагели хорошо поглощают водяные пары при очень низкой относительной влажности воздуха. Сорбция
|
Таблица 1.L Характеристики силикагелей |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
19 |
ББК 20.1 Р85
УДК 543.27 (031)
Рецензент: канд. техн. наук В.А.Симонов
Руководство по контролю вредных веществ в воздухе рабочей зоны: Справ. изд./С.И.Муравьева, М.И.Буковский, Е.К.Прохорова и др. — М.: Химия, 1991.368 с.
ISBN 5-7245-0141-4
О писаны методы отбора проб и воздухозаборная аппаратура, способы извлечения, разделения и идентификации химических веществ. Даны краткие технические характеристики отечественных аспирационных устройств, поглотительных сосудов. Изложены статические и динамические методы приготовления смесей вредных веществ с воздухом. Приведены современные инструментальные методы анализа. Даны краткие характеристики отечественных и зарубежных индикаторных трубок. Рассмотрены автоматические газоанализаторы.
Книга предназначена для химиков лабораторий промышленных предприятий, научно-исследовательских институтов и других организаций, занимающихся разработкой методов контроля состояния воздушной среды производственных помещений.
ББК 20.1
1502020000-045 050(01)—91
Справочное издание
Муравьева Софья Ивановна, Буковский Михаил Иванович,
Прохорова Евгения Константиновна, Бабина Мария Дмитриевна,
Колосник Михаил Иосифович
РУКОВОДСТВО ПО КОНТРОЛЮ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ РАБОЧЕЙ ЗОНЫ Редактор В.Л.Абрамоаа Художник Е.В.Бехетоа Художественный редактор КДС.Федоров Технический редактор Л.Н.Богдакова Корректор Л.В.Лазуткина
ИБ N° 2484
Сдано в набор 9.08.90 г. Подписано в печать 23.07.91 г. Формат 60×88 1/16.
Бум. офсетная № 2. Гарнитура Литературная. Печать офсетная.
Усл.печ.л. 22,54. Усллср.-отт. 22,54. Уч.-изд.л. 26,68.
Тираж 19200 экз. Заказ 1241 Цена 3 р.
Ордена «Знак Почета»кэдательство «Химия».
107076, Москва, Стромынка, 21, корп. 2 Диапозитивы изготовлены в МП «Измайлово»
105856 ГСП, Москва Е-37, гор. им. Баумана.
Отпечатано с готовых пленок Московской тип. N9 11 113105, Москва, Нагатинская ул., д. 1.
ISBN 5-7245-0141-4 © С. И. Муравьева, M.HEj копский,
Е. К. Прохорова, М. Д. Бабина, М. И. Колесник, 1991
при отборе проб на микропористые силикагели.
|
Тип |
Размер пор. мм |
Насыпная плот- Плотность, кг/м3 ность,кг/м |
Эффективный диаметр пор, нм |
||
|
СССР |
|||||
|
СаХ |
0,6—0,24 |
0,48—0,75 |
750 — 1000 |
— |
|
|
NaX |
0,6—0,24 |
0,48—0,75 |
750—1000 |
— |
|
|
США |
|||||
|
ЗА |
1,5 |
752 |
1000—1100 |
0,3 |
|
|
4А |
1,5 и 3,0 |
721 |
1100 |
0,4 |
|
|
5А |
1,5 и 3,0 |
— |
1150 |
0,5 |
|
|
10А |
— |
576 |
900—1000 |
0,8 |
|
|
13Х |
— |
576 |
940 |
0,9 |
|
|
водяного |
пара |
приводит |
к вытеснению |
средних |
и неполярных |
|
веществ |
при п |
робоотборе, |
, что является |
главным |
препятствием |
Способность силикагеля катализировать некоторые реакции (дегидратации, конденсации, полимеризации) ограничивает срок хранения отобранных проб. Однако при низких температурах (—5 “ — 10°С) сохранность проб значительно увеличивается. При хранении силикагели изменяют свои свойства. Свежеприготовленный силикагель стареет через 1—2 года, при этом частично изменяется и его сорбционная активность.
Молекулярные сита. Молекулярные сита или цеолиты — синтетические сорбенты со строго определенным размером пор в кристаллической решетке. Они состоят из кремний-алюмо-кис-лородных кубооктаэдров, имеющих формулу
ЫагО- АЬОз- nSiCb • Н2О. Наиболее широкое применение нашли два основных кристаллографических типа А и X, отличающихся по молярному замещению отдельных компонентов и по кристаллизационному скелету. У типа А молярное соотношение БЮггАЬОзСл) колеблется в пределах 1,87—2,0, а у типа X в пределах 2,3—3,3. Скелет молекулярных сит состоит из кубооктаэдров, содержащих 24 тетраэдра. Тетраэдр построен так, что ионы кремния и алюминия находятся в центре, а ионы кислорода на вершинах. Полости молекулярных сит возникают путем соединения кубооктаэдров, образуя пространственный скелет с помощью кислородных связей. Катионы натрия связаны с помощью йодной связи и легко подвергаются ионообмену, в результате чего образуются одновалентные ионы (калий, серебро) или ионы щелочноземельных металлов (кальций). При таком ионо-обмене алюмо-силикатный скелет сохраняется, однако изменяется диаметр у входа в поры в результате различных радиусов катионов, что приводит к образованию ситового эффекта.
Основные типы сит 4А и 5А, так же как 10Х и 13Х различаются только катионом кристаллической решетки. Поры молекул яр-
20
ОГЛАВЛЕНИЕ
Предисловие………………….6
Введение …………………. 8
Глава /.Отбор проб воздуха……………9
1.1. Отбор проб в жидкие среды………..15
1.2. Отбор проб на твердые сорбенты……….17
1.3. Хемосорбция………………30
1.4. Отбор проб в охлаждаемые ловушки………31
1.5. Отбор проб в сосуды ограниченной вместимости…..32
1.6. Отбор проб на фильтры…………..32
1.7. Индивидуальная активная дозиметрия ………36
1.8. Индивидуальная пассивная дозиметрия……..43
1.8.1. Аналитические методы контроля, применяемые при
пассивном отборе проб воздуха …….. 52
1.8.2. Влияние окружающей среды на адсорбцию веществ в
пассивном дозиметре………….53
1.8.3. Установление скорости поглощения вещества пассивными дозиметрами………….55
1.9. Расчет концентрации веществ…………58
Глава 2. Аппаратура для отбора проб воздуха……..62
2.1. Побудители расхода……………63
2.2. Расходомеры………………65
2.3. Аспирационные устройства…………67
Глава 3. Извлечение, концентрирование и идентификация веществ . . 75
3.1. Извлечение и концентрирование веществ…….75
3.2. Идентификация веществ………….87
Г лава 4. Методы приготовления смесей вредных веществ с воздухом . . 92
4.1. Статические методы……………94
4.2. Динамические методы…………..96
4.3. Установки, используемые в практике контроля воздуха … 99
4.4. Приготовление смесей аэрозолей с воздухом…..104
Глава 5. Физико-химические методы анализа………106
5.1. Газовая хроматография………….106
5.1.1. Хроматограмма и ее параметры……..107
5.1.2. Хроматографические колонки и коэффициент разделения 109
5.1.3. Выбор неподвижной фазы и условий разделения . .110
5.1.4. Твердые носители для газожидкостной хроматографии
и адсорбенты для газоадсорбционной хроматографии 113
5.1.5. Детекторы……………. 116
5.1.6. Качественный анализ…………118
5.1.7. Количественный анализ………..118
5.1.8. Хроматографы……………119
5.1.9. Дозаторы для ввода проб в хроматограф . . . .124
5.1.10. Приготовление сорбентов………..125
5.2. Тонкослойная хроматография………..126
5.2.1. Адсорбционная хроматография………127
3
5.2.2. Распределительная хроматография…….128
5.2.3. Сорбенты для ТСХ…………139
5.2.4. Пластинки для ТСХ…………133
5.2.5. Процесс разделения веществ на пластинке . . . .135
5.2.6. Обнаружение веществ на хроматограмме…..136
5.2.7. Количественный анализ………..138
5.2.8. Оборудование для ТСХ…………139
5.3. Жидкостная хроматография…………139
5.4. Ионная хроматография………….149
5.5. Масс-спектрометрия и хромато-масс-спектрометрия …. 153
5.6. Фотометрия………………157
5.7. Спектроскопические методы…………171
5.7.1. Атомно-эмиссионная спектрометрия……..172
5.7.2. Атомно-абсорбционная спектрометрия…….175
5.7.3. Атомно-флуоресцентная спектрометрия……178
5.7.4. Оптико-акустическая спектрометрия…….180
5.8. Вольтамперометрия……………184
5.9. Ионометрия………………188
Г лава б. Метрологическое обеспечение контроля воздуха…..193
Глава 7. Измерение концентрации вредных веществ индикаторными
трубками……………….202
7.1. Основные принципы работы индикаторных трубок…..203
7.2. Подготовка и проведение измерений индикаторными трубками 208
7.3. Воздухозаборные устройства для индикаторных трубок . . 209
7.3.1. Аспиратор сильфонный АМ-5………211
7.3.2. Воздухозаборное устройство газоанализатора УГ-2 . . 215
7.3.3. Ручной поршневой насос Китагавы…….220
7.4. Г азоопределители химические ГХ-М………221
7.5. Газоанализатор универсальный переносной УГ-2 …. 227
7.5.1. Комплекты индикаторных средств……..231
7.5.2. Подготовка и проведение анализа воздушной среды
газоанализатором УГ-2…………237
7.6. Зарубежные средства индикации……….242
7.7. Индикаторные трубки длительного действия для определения
среднесменных концентраций вредных веществ в воздухе . .251
Глава 8. Автоматический анализ воздушной среды производственных
помещений……………… 254
8.1. Общие сведения…………….254
8.2. Автоматические сигнализаторы довзрывоопасных концентраций ………………..255
8.3. Газоанализаторы вредных веществ в воздухе……263
8.4. Магнитные и термомагнитные газоанализаторы кислорода . . 277
8.5. Общие требования к выбору мест отбора проб воздуха и
к установке датчиков автоматических анализаторов контроля воздушной среды…………….279
8.6. Зарубежные средства автоматического контроля воздушной
среды………………..287
Г лава 9. Универсальная система химического анализа воздуха рабочей
зоны…………………296
Глава 10. Комплектные лаборатории, выпускаемые отечественной промышленностью ……………..310
4
Приложения………………….314
1. Эмпирические коэффициенты для расчета давления пара
индивидуальных веществ в зависимости от температуры и летучести веществ при 20°С…………314
2. Коэффициенты К для приведения объема воздуха к стандартным условиям……………320
3. Перечень Методических указаний (МУ) по измерению концентраций вредных веществ в воздухе рабочей зоны, утвержденных М3 СССР и опубликованных в сборниках …. 321
4. Перевод объемной концентрации (1:1000 ООО, 1 ррт) в массовую (мг/л) для газов и паров……….339
5. Перечень веществ, контролируемых термохимическими и
искровыми сигнализаторами ………..345
6. Перечень веществ, контролируемых сигнализатором СТХ-7 345
7. Перечень веществ, контролируемых сигнализатором СДК-3 345
8. Методика определения количества и мест установки пер
вичных преобразователей информации (датчиков) о составе воздушной среды помещений в системах взрывопредупреж-дения ……………….348
Библиографический список……………..354
ПРЕДИСЛОВИЕ
Рост и интенсификация всех отраслей промышленности приводит к значительному загрязнению химическими веществами среды обитания человека, увеличению контакта с ними в процессе производства и в быту. В связи с этим особую значимость приобретает контроль объектов окружающей среды и в первую очередь производственного воздуха. В решении этой проблемы важную роль играет прежде всего знание вида и концентрации присутствующих в воздухе загрязнителей, обнаружение и учет которых осуществляется методами химического анализа.
За последние годы промышленно-санитарная химия значительно продвинулась вперед в области качественного и количественного анализа производственного воздуха. Для обнаружения химических загрязнителей в воздухе и других средах стали широко применять современные физико-химические методы анализа: газовую, тонкослойную и жидкостную хроматографию, атомно-абсорбционную и ультрафиолетовую спектрофотометрию, ионометрию, хромато-масс-спектрометрию. Это позволило санитарно-промышленной химии сделать значительный скачок в области анализа микропримесей химических загрязнителей. Благодаря использованию современных приборов и аппаратуры, выпускаемых отечественной промышленностью, стало возможно проводить анализ сложных многокомпонентных смесей неизвестного состава и устанавливать количественное содержание каждого компонента в отдельности. Накопился огромный методический материал в области санитарно-химического анализа, который необходимо обобщить и опубликовать.
С момента выхода в свет фундаментального руководства по промышленно-санитарной химии «Методы определения вредных веществ в воздухе» под редакцией О.Д. Хализовой прошло уже более 20 лет. За эти годы вышли в свет книги «Химический анализ воздуха промышленных предприятий», авторы Е.А. Перегуд и Е.В. Гернет, «Химический анализ воздуха», автор Е.А. Перегуд. В эти руководства включены методики определения отдельных химических веществ в воздухе, преимущественно колориметрические и фотометрические, первые из которых в настоящее время не рекомендуются для использования. Заслуживает внимания книга В. Лейта «Определение загрязнений воздуха в атмосфере и на рабочем месте», изданная в 1980 г.
В 1982 г. вышло новое руководство — «Санитарно-химический контроль на промышленных предприятиях» под ред. С.И. Муравьевой. В книге изложены общие и частные вопросы по промышленно-санитарной химии. В настоящее время эта книга является
настольным руководством для научных и практических работников.
В 1988 г. издан «Справочник по контролю вредных веществ в воздухе», авторы С.И. Муравьева, Н.И. Казнина, Е.К. Прохорова. В нем в краткой форме представлены основные аспекты санитарного контроля воздуха и в аннотированной форме изложены методики определения неорганических и органических соединений в воздухе рабочей зоны и в атмосфере. В этом же году издана книга Симонова и др. «Анализ воздушной среды при переработке полимерных материалов».
Подготавливаемое к изданию руководство по контролю вредных веществ в воздухе будет состоять из трех томов. В 1-м томе настоящего руководства подробно освещены основные проблемы отбора проб воздуха, способы извлечения, разделения и идентификации компонентов проб, концентрирование, а также количественное определение и метрологическое обеспечение. Наряду с этим описаны новые способы отбора проб воздуха — индивидуальная активная и пассивная дозиметрия. Широко представлены современные инструментальные методы анализа применительно к промышленно-санитарной химии: газовая и жидкостная хроматография, масс-спектрометрия для разделения сложных газовых смесей неизвестного состава, вольтамперометрия, УФ- и видимая спектрофотометрия, атомно-абсорбционная спектрофотометрия, ионометрия и др. Большое внимание уделено описанию экспрессных методов с применением индикаторных трубок отечественного и зарубежного производства, а также газоанализаторам, позволяющим непосредственно на рабочем месте устанавливать уровни загрязнения воздуха токсическими веществами.
Настоящее руководство будет полезно для специалистов, работающих в области промышленно-санитарного анализа в научно-исследовательских институтах, на санитарно-эпидемиологических станциях, а также в заводских лабораториях.
Предисловие, введение, гл. 1, 3, 5 (разд. 5.1 —5.3, 5.5, 5.6), приложения 1—5 написаны С.И. Муравьевой и М.Д. Бабиной; гл. 2, 4, 5 (разд. 5.4, 5.7 — 5.9), гл. 6, 9 написаны Е.К. Прохоровой; гл. 7, 8, приложения 6—16 написаны М.И. Буковским и М.И. Колесником; гл. 10 написана М.Д. Бабиной. В составлении библиографического списка участвовали все авторы.
ВВЕДЕНИЕ
В нашей стране большое внимание уделяется вопросам оздоровления условий труда. Еще в 30-е годы был введен предупредительный санитарный надзор за внедрением в промышленность новых химических веществ. Новые химические вещества внедряются в производство после глубокого изучения их токсических свойств. В законодательном порядке устанавливаются предельно допустимые концентрации вредных веществ в воздухе рабочей зоны и ориентировочно безопасные уровни, что способствует оздоровлению условий труда на производстве.
Решение этих важных задач, а также глубокие исследования биологического действия химических соединений на человека и животных невозможны без наличия системы контроля, главной составной частью которого является анализ вредных веществ в производственной атмосфере. Для проведения таких исследований необходимы надежные физико-химические методы количественного определения химических веществ, отвечающие гигиеническим требованиям.
Для адекватной оценки воздушной среды с учетом конкретных производственных факторов (температуры и влажности воздуха, наличия сопутствующих примесей и других) применяют методы, которые должны отвечать установленным требованиям. До недавнего времени указанные требования не были регламентированы. В большинстве случаев в методах контроля вообще отсутствовали сведения о точности и мешающем влиянии сопутствующих примесей. Не была также регламентирована длительность отбора проб в воздухе в случае измерения концентраций вредных веществ при установлении их соответствия предельно допустимым уровням. Отсутствие указанных сведений крайне затрудняло объективную оценку и выбор наиболее надежных методов, а также не позволяло достаточно обоснованно судить о правильности измерения.
Совместными усилиями химиков, гигиенистов и токсикологов проведена регламентация основных требований к методам контроля по чувствительности (нижнего предела определяемых содержаний), точности и селективности, времени отбора проб воздуха.
Разработанные требования к методам определения химических веществ в воздухе рабочей зоны [1, 2] должны обеспечивать избирательное определение вредного вещества в воздухе на уровне 1/2 ПДК в присутствии сопутствующих примесей с суммарной погрешностью, не превышающей ±25% [1, 2]. При выборе методов количественного определения вредных веществ необходимо пользоваться Методическими указаниями по измерению концентраций вредных веществ в воздухе рабочей зоны, утвержденными М3 СССР, которые выходят в свет отдельными сборниками. Методические указания утверждены более чем для 1300 химических веществ.
8
Глава 1 ОТБОР ПРОБ ВОЗДУХА
Существенным этапом санитарно-химических исследований воздушной среды является отбор пробы воздуха для определения содержания микропримесей токсичных соединений. Результаты самого точного и тщательно выполненного анализа теряют смысл в случае неправильной подготовки к отбору пробы и неверного его выполнения. Поэтому при разработке методов контроля этому этапу уделяют большое внимание.
Пробы воздуха следует отбирать на местах постоянного и временного пребывания работающих, при характерных производственных условиях с учетом особенностей технологического процесса (непрерывный, периодический), температурного режима, количества выделяющихся химических веществ; физико-химических свойств контролируемых веществ, их агрегатного состояния в воздухе, летучести, давления паров и возможности их превращения (окисление, гидролиз, деструкция и др.); температуры и влажности окружающей среды; класса опасности и биологического действия химического соединения.
При наличии в воздухе нескольких химических веществ или сложных многокомпонентных смесей неизвестного состава необходимо предварительно провести идентификацию смесей и определить приоритетные — наиболее опасные и характерные компоненты, на которые следует ориентироваться при оценке состояния воздушной среды.
Контроль за соблюдением максимально-разовой (м.р.) ПДК и ОБУВ проводят при непрерывном или последовательном отборе в течение 15 мин в любой точке рабочей зоны при условии достижения предела обнаружения определяемого вещества. Если предел обнаружения метода анализа дает возможность в течение 15 мин отобрать не одну, а несколько проб воздуха, то нужно определить среднее значение из результатов отобранных проб за указанный период времени. Если данным методом невозможно обнаружить вещество на уровне 0,5 ПДК м.р. за 15 мин, допускается увеличение продолжительности отбора проб до 30 мин.
Если стадия технологического процесса настолько коротка, что нельзя отобрать в одну пробу необходимое для анализа количество вещества, то отбор проб в эту же концентрационную трубку (фильтр) или поглотительный прибор необходимо продолжить при повторении операции.
При санитарно-гигиенических исследованиях производственной атмосферы с длительными стадиями технологического процесса отбор проб необходимо проводить с учетом начала, сере-
9
Текст ГОСТ Р 59670-2021 Воздух рабочей зоны. Общие требования к методикам определения содержания химических веществ
ГОСТ Р 59670-2021
(ИСО 20581:2016)
НАЦИОНАЛЬНЫЙ СТАНДАРТ РОССИЙСКОЙ ФЕДЕРАЦИИ
ВОЗДУХ РАБОЧЕЙ ЗОНЫ
Общие требования к методикам определения содержания химических веществ
Workplace air. General requirements for the performance of procedures for the measurement of chemical agents
ОКС 13.040.30
Дата введения 2022-01-01
Предисловие
1 ПОДГОТОВЛЕН Акционерным обществом «Научно-исследовательский институт охраны атмосферного воздуха» (АО «НИИ Атмосфера») на основе собственного перевода на русский язык англоязычной версии стандарта, указанного в пункте 4
2 ВНЕСЕН Техническим комитетом по стандартизации ТК 457 «Качество воздуха»
3 УТВЕРЖДЕН И ВВЕДЕН В ДЕЙСТВИЕ Приказом Федерального агентства по техническому регулированию и метрологии от 3 сентября 2021 г. N 913-ст
4 Настоящий стандарт является модифицированным по отношению к международному стандарту ИСО 20581:2016* «Воздух рабочей зоны. Общие требования к методикам определения содержания химических веществ» (ISO 20581:2016 «Workplace air — General requirements for the performance of procedures for the measurement of chemical agents», MOD) путем включения дополнительного примечания в раздел 6 и новых библиографических ссылок для учета особенностей национальной стандартизации, которые выделены в тексте курсивом, а объяснение причин их включения приведено в сносках.
________________
* Доступ к международным и зарубежным документам, упомянутым в тексте, можно получить, обратившись в Службу поддержки пользователей.
Сведения о соответствии ссылочных национальных и межгосударственных стандартов международным и европейским стандартам, использованным в качестве ссылочных в примененном международном стандарте, приведены в дополнительном приложении ДА
5 ВЗАМЕН ГОСТ Р ЕН 482-2012
6 ПЕРЕИЗДАНИЕ. Март 2022 г.
Правила применения настоящего стандарта установлены в статье 26 Федерального закона от 29 июня 2015 г. N 162-ФЗ «О стандартизации в Российской Федерации». Информация об изменениях к настоящему стандарту публикуется в ежегодном (по состоянию на 1 января текущего года) информационном указателе «Национальные стандарты», а официальный текст изменений и поправок — в ежемесячном информационном указателе «Национальные стандарты». В случае пересмотра (замены) или отмены настоящего стандарта соответствующее уведомление будет опубликовано в ближайшем выпуске ежемесячного информационного указателя «Национальные стандарты». Соответствующая информация, уведомление и тексты размещаются также в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет (www.rst.gov.ru)
Введение
В национальном законодательстве и нормативных правовых актах установлены требования в отношении оценки потенциального воздействия на работника химических веществ, находящихся в воздухе рабочей зоны. Одним из способов оценки такого воздействия является измерение концентрации химического вещества в воздухе в зоне дыхания работника. Применяемые при таких измерениях методики должны обеспечивать получение надежных и достоверных результатов для цели сравнения результатов измерений воздействия с предельными значениями профессионального воздействия и для обеспечения приемлемых стратегий контроля.
Настоящий стандарт вводит общие требования для выполнения методик измерений в процессе количественной оценки воздействия. Для различных типов методик измерений и измерительных устройств
подготовлены специальные международные и европейские стандарты. К ним относятся стандарты, рассматривающие пробоотборники частиц в воздухе (ГОСТ Р ЕН 13205), диффузионные пробоотборники (ГОСТ Р ИСО 16107 и ГОСТ Р ЕН 838), пробоотборники с прокачкой пробы
[1]
, индикаторные трубки
[2]
, насосы для индивидуальных пробоотборников (ГОСТ Р ИСО 13137), металлы и металлоиды в частицах, находящихся в воздухе
[3]
, смеси частиц и пара в воздухе
[4]
и приборы для прямого определения токсичных газов и паров
[5]
. Методы измерений и требования к средствам измерений установлены этими стандартами таким образом, чтобы обеспечить возможность соблюдения требований настоящего стандарта. При отсутствии стандарта на методы измерений для конкретного химического вещества следует руководствоваться общими требованиями настоящего стандарта.
________________
Под измерительными устройствами следует понимать средства измерений, измерительные системы, а также оборудование для отбора проб.
Требования к характеристикам эффективности, приведенные в настоящем стандарте, предназначены для применения в условиях окружающей среды на рабочем месте. Ввиду широкого диапазона условий окружающей среды настоящий стандарт определяет те требования, которые должны быть обязательно соблюдены с учетом методик измерений при проведении испытаний в предписанных лабораторных условиях.
Пользователь отвечает за выбор тех методик измерений или измерительных устройств, которые должны соответствовать требованиям настоящего стандарта. Одним из способов этого выбора является получение информации или подтверждения от разработчика методики или изготовителя устройства. Типовые испытания или, в более общем случае, оценка характеристик эффективности методик измерений или измерительных устройств могут быть проведены изготовителем, пользователем, испытательной лабораторией или, что наиболее целесообразно, научно-исследовательской лабораторией. Ряд существующих методик измерений, применяемых для измерений показателей воздуха рабочей зоны, были испытаны только в части минимально требуемого диапазона измерений, но не во всем диапазоне, либо не без учета предмета влияния всех факторов окружающей среды и потенциальных мешающих веществ. Если эти частично валидированные методики измерений соответствуют требованиям к характеристикам эффективности, приведенным в настоящем стандарте, то они могут быть применены в настоящее время. Тем не менее испытания следует проводить во всех диапазонах с учетом обоснованности их применения на практике.
1 Область применения
Настоящий стандарт определяет общие требования к характеристикам эффективности методик определения содержания химических веществ в воздухе рабочей зоны. Данные требования применяются ко всем этапам методик измерений независимо от физической формы химического вещества (газ, пар, частицы в воздухе), а также к методикам с раздельным отбором проб и аналитическими методами и к методикам устройств прямого определения.
Настоящий стандарт определяет требования, которые должны быть обязательно выполнены при испытании методик измерений в лабораторных условиях в широком диапазоне условий окружающей среды.
2 Нормативные ссылки
В настоящем стандарте использованы нормативные ссылки на следующие стандарты:
ГОСТ 34100.3-2017 Неопределенность измерения. Часть 3. Руководство по выражению неопределенности измерения
ГОСТ Р 8.563 Государственная система обеспечения единства измерений. Методики (методы) измерений
ГОСТ Р ИСО 7708 Качество воздуха. Определение гранулометрического состава частиц при санитарно-гигиеническом контроле
ГОСТ Р ИСО 13137 Воздух рабочей зоны. Насосы для индивидуального отбора проб химических и биологических веществ. Требования и методы испытаний
ГОСТ Р ИСО 16107 Воздух рабочей зоны. Оценка характеристик диффузионных пробоотборников
ГОСТ Р ЕН 838 Воздух рабочей зоны. Диффузионные пробоотборники, используемые при определении содержания газов и паров. Требования и методы испытаний
ГОСТ Р ЕН 13205 Воздух рабочей зоны. Оценка характеристик приборов для определения содержания твердых частиц
ГОСТ Р ИСО 15767 Воздух рабочей зоны. Контроль и оценка неопределенности взвешивания проб аэрозолей
ГОСТ Р ИСО 21748 Статистические методы. Руководство по использованию оценок повторяемости, воспроизводимости и правильности при оценке неопределенности измерений
Примечание — При пользовании настоящим стандартом целесообразно проверить действие ссылочных стандартов в информационной системе общего пользования — на официальном сайте Федерального агентства по техническому регулированию и метрологии в сети Интернет или по ежегодному информационному указателю «Национальные стандарты», который опубликован по состоянию на 1 января текущего года, и по выпускам ежемесячного информационного указателя «Национальные стандарты» за текущий год. Если заменен ссылочный стандарт, на который дана недатированная ссылка, то рекомендуется использовать действующую версию этого стандарта с учетом всех внесенных в данную версию изменений. Если заменен ссылочный стандарт, на который дана датированная ссылка, то рекомендуется использовать версию этого стандарта с указанным выше годом утверждения (принятия). Если после утверждения настоящего стандарта в ссылочный стандарт, на который дана датированная ссылка, внесено изменение, затрагивающее положение, на которое дана ссылка, то это положение рекомендуется применять без учета данного изменения. Если ссылочный стандарт отменен без замены, то положение, в котором дана ссылка на него, рекомендуется применять в части, не затрагивающей эту ссылку.
3 Термины и определения
В настоящем стандарте применены термины и определения в соответствии с [6].
ИСО и МЭК также содержат терминологические базы данных для использования в стандартизации по следующим адресам:
— платформа онлайн-просмотра ИСО, которая доступна на https://www.iso.org/obp;
— электропедия МЭК, которая доступна на http://www.electropedia.org/.
4 Классификация
4.1 Общие положения
В настоящем стандарте измерения классифицированы в соответствии с целями их выполнения. Эта классификация основана на ранее принятой измерительной стратегии.
4.2 Скрининговые измерения
средневзвешенной по времени концентрации
________________
Скрининговыми измерениями называют измерения, используемые для выявления, идентификации и полуколичественной оценки содержания аналита в пробе.
Скрининговые измерения для определения средневзвешенной по времени концентрации проводят для получения полуколичественной информации об уровнях воздействия. Такая информация используется для выявления потенциальных опасностей для здоровья и оценки риска для здоровья на основе вероятной серьезности вреда и вероятности его возникновения. С их помощью этих измерений можно определить, является ли воздействие значительно ниже или выше предельного значения профессионального воздействия (ПЗПВ)
. Применяют аппаратуру, предназначенную для обнаружения и измерения концентрации химических веществ, которая отвечает требованиям настоящего стандарта и определяет средневзвешенное по времени значение.
________________
К ПЗПВ следует относить гигиенические нормативы: предельно допустимая концентрация (ПДК) и ориентировочные безопасные уровни воздействия (ОБУВ).
4.3 Скрининговые измерения изменения концентрации во времени и/или в пространстве
Скрининговые измерения изменения концентрации химического вещества во времени и (или) в пространстве используют для получения общего представления о потенциальных распределениях в воздухе рабочей зоны для определения мест и периодов ее повышенного значения. Скрининговые измерения также предоставляют информацию о местонахождении и интенсивности источников выбросов и позволяют оценить эффективность вентиляции или других технических мер. Применяют аппаратуру, предназначенную для обнаружения и измерения концентрации химических веществ, которая соответствует требованиям настоящего стандарта.
4.4 Измерения для сравнения с предельным значением профессионального воздействия
Измерения могут быть проведены для сравнения с ПЗПВ при условии, что лежащий в их основе метод соответствует критерию принятой неопределенности. Цель таких измерений может состоять в проверке того, не превышены ли ПЗПВ на рабочем месте в данный момент времени, а также вследствие значительных изменений в условиях труда, технологических процессах, производимой продукции, применяемых химикатах или после изменения ПЗПВ.
4.5 Периодические измерения
Периодические измерения используют для определения изменений условий воздействия с момента проведения измерений для сравнения с ПЗПВ или эффективности мер контроля. Интервал между измерениями должен быть установлен на основе первоначальной оценки профессионального воздействия или последующих поправок к ней.
Примечание — При условии, что состав химических веществ в воздухе рабочей зоны определен при первоначальной оценке содержания вредных веществ и не изменяется со временем, для последующих периодических измерений допускается использовать методики с пониженной селективностью.
5 Требования к эффективности
5.1 Общая информация
Требования эффективности методик измерений зависят от цели измерений. Так как требования эффективности скрининговых измерений менее строгие по отношению к периодическим измерениям и измерениям для сравнения с ПЗПВ, они приведены в 5.2 и 5.3 в общем виде.
5.2 Скрининговые измерения средневзвешенной по времени концентрации
Обоснование цели этих измерений приведено в 4.2. Методика данного вида измерений должна обладать:
a) адекватной селективностью для конкретного химического вещества;
b) периодом усреднения, продолжительность которого меньше или равна продолжительности регламентированного периода, установленного для ПЗПВ;
c) диапазоном измерений, включающим значение ПЗПВ;
d) расширенной неопределенностью, соответствующей поставленной цели.
5.3 Скрининговые измерения изменения концентрации во времени и/или в пространстве
Основание цели этих измерений приведено в 4.3. Методика данного вида измерений должна обладать:
a) адекватной селективностью для конкретного химического вещества;
b) коротким периодом усреднения (при определении изменения концентрации во времени
5 мин; в пространстве —
15 мин);
c) диапазоном измерений, соответствующим цели их проведения (см. 4.3);
d) расширенной неопределенностью, соответствующей поставленной цели.
5.4 Измерения для сравнения с предельным значением профессионального воздействия и периодические измерения
5.4.1 Однозначность
Методика измерений должна обеспечивать получение однозначного результата измерения концентрации конкретного химического вещества в установленном диапазоне измерений, т.е. полученная аналитическим путем оценка должна включать в себя одно числовое значение концентрации химического вещества с известной неопределенностью.
5.4.2 Селективность
Методика измерений должна содержать соответствующую информацию о природе и значимости любых веществ, влияющих на достоверность результатов измерений.
Примечание — Требования к селективности методик отличаются в зависимости от имеющейся информации о составе воздуха рабочей зоны. Если заранее неизвестно обо всех загрязнителях, присутствующих в воздухе, то методика измерений должна иметь высокую селективность. При наличии данных о том, какие загрязнители обнаружены в воздухе, при отсутствии мешающих веществ можно использовать методику измерений с низкой или адекватной селективностью.
Если ПЗПВ установлено для определенной фракции частиц (см. ГОСТ Р ИСО 7708), то методика измерений содержания химического вещества, присутствующего в воздухе в виде частиц, должна предусматривать отбор проб указанной фракции.
Примечание — В дополнение к пробоотборникам, отвечающим требованиям ГОСТ Р ИСО 7708, существуют пробоотборники с встроенным расходомером (см. ГОСТ Р ЕН 13205).
Если для различных разновидностей одного и того же химического соединения установлены различные ПЗПВ, то методика измерений должна обеспечивать определение соответствующих индивидуальных разновидностей соединения.
5.4.3 Период усреднения
Период усреднения равен продолжительности отбора проб, т.е. не более регламентированного периода, для которого установлено ПЗПВ. В зависимости от методов отбора проб, период отбора может варьироваться.
Примечание — Средняя концентрация за полную смену, обычно 8-часовая взвешенная по времени концентрация, дает репрезентативное описание ситуации профессионального воздействия на работника.
Пиковые значения концентрации вредного вещества, которые могут наблюдаться в течение рабочей смены систематически или случайным образом, не должны превышать ПЗПВ, установленного для краткосрочного периода.
5.4.4 Диапазон измерений
Диапазон измерений методики, указанной в таблице 1, должен включать, по крайней мере, концентрации от 0,1 до 2 ПЗПВ для долговременных измерений и от 0,5 до 2 ПЗПВ — для кратковременных. Соответствующее ПЗПВ должно быть определено соответствующими органами при использовании установленной методики измерений.
Примечание — Некоторые ПЗПВ в воздухе рабочей зоны приведены в [11].
5.4.5 Расширенная неопределенность
Требования к расширенной неопределенности приведены в таблице 1.
Таблица 1 — Требования к расширенной неопределенности при проведении измерений для сравнения с ПЗПВ и при периодических измерениях
|
Регламентированный период |
Диапазон измерений |
Относительная расширенная неопределенность, % |
Относительная расширенная неопределенность (смеси пара и частиц в воздухе), % |
|
Краткосрочный (например, 15 мин) |
От 0,5 до 2 ПЗПВ |
50 |
50 |
|
Долгосрочный |
От 0,1 до 0,5 ПЗПВ |
50 |
50 |
|
Долгосрочный |
Св. 0,5 до 2 ПЗПВ |
30 |
50 |
Примечание — Вариация воздействия химических веществ на рабочем месте может быть значительно больше, чем указано в неопределенности единичного измерения, рассчитанной в соответствии с настоящим стандартом. Это связано с временной и пространственной изменчивостью воздействия на рабочем месте.
5.4.6 Химические вещества, для которых отсутствуют соответствующие методы
Предельные значения определяются регулирующими органами независимо от позиции разработчиков методики измерений. Если для установленного предельного значения отсутствует методика измерений, удовлетворяющая требованиям 5.4.4 и 5.4.5, то применяют методику измерений, характеристики которой максимально близки к указанным в этих пунктах. Используемый метод должен четко определять ограничения при измерении на пределе воздействия.
5.5 Многоэтапные методики
Требования эффективности, установленные в 5.2-5.4, относятся ко всей методике измерений в целом, даже если она состоит из нескольких отдельных этапов, таких как подготовка оборудования, отбор проб, их транспортирование и хранение, подготовка проб и анализ. Допускается оценивать неопределенность для каждого этапа отдельно с последующим объединением оценок в общую неопределенность измерений.
5.6 Транспортирование, обработка и/или хранение
Транспортирование, обработку и/или хранение проб, при необходимости, следует осуществлять таким образом, чтобы сохранялась физическая и химическая целостность проб в период между отбором проб и анализом.
5.7 Условия окружающей среды
Испытание в отношении влияния условий окружающей среды (например, температуры, влажности, атмосферного давления и/или скорости воздуха) на характеристики эффективности методики должно быть проведено в лаборатории. Требования эффективности, затрагивающие однозначность, селективность, расширенную неопределенность, минимальный диапазон измерений и период усреднения, должны быть соблюдены для всех условий, которые установлены на рабочем месте.
Примечание — Проводить полную оценку влияния условий внешней среды непосредственно на рабочих местах нецелесообразно с практической точки зрения ввиду значительной стоимости и больших затрат времени, поэтому согласно настоящему стандарту такую оценку получают моделированием внешних факторов в условиях лаборатории. Вместе с тем, проверка методики измерений на рабочем месте позволяет получить ценную дополнительную информацию (например, о влиянии мешающих веществ, образующихся на соседних рабочих местах).
В методике измерений должен быть указан диапазон изменения факторов окружающей среды, в котором обеспечивается выполнение требований по 5.2-5.5.
5.8 Описание методики измерений
Оформление методики измерений в документальном виде осуществляют в соответствии с [7]. Актуальность приведения или исключения пунктов или подпунктов применяемой методики определена в каждом конкретном случае отдельно. Методика измерений должна содержать всю информацию, необходимую для проведения измерений, включая сведения о достижимой расширенной неопределенности, диапазоне измерений, периоде усреднения, мешающих веществах, условиях окружающей среды и других факторах, которые могут повлиять на эффективность методики измерений.
Если применяют поправочные коэффициенты, например в случае реального и объяснимого смещения, связанного с влиянием окружающей среды, то они должны быть обоснованы в методике измерений.
Примечание — Пример структуры описания методики измерений, основанный на [7], приведен в приложении A.
5.9 Размерность результата
Окончательный результат измерений должен быть выражен в таких же единицах, как и ПЗПВ, что можно достичь за счет получения результата непосредственно в выбранных единицах или посредством соответствующего пересчета.
Это требование не является обязательным для скрининговых измерений изменения концентрации во времени и/или в пространстве.
5.10 Дополнительные требования
В дополнение к требованиям, приведенным в 5.2-5.9, для некоторых методик и измерительных устройств при необходимости следует выполнять дополнительные требования, установленные в ГОСТ Р ИСО 13137, ГОСТ Р ИСО 16107, ГОСТ Р ЕН 838, ГОСТ Р ЕН 13205, а также в [1]-[5].
В таблице 2 приведены дополнительные требования к параметрам, подлежащим испытаниям.
Таблица 2 — Дополнительные требования
|
Стандарт |
Необходимые параметры, подлежащие испытаниям |
|
ГОСТ Р ИСО 13137 |
Характеристика и масса насосов, безопасность конструкции, время работы, пуск и длительная работа, кратковременное прерывание воздушного потока, механическая прочность, точность таймера, электромагнитная совместимость, взрывоопасность, зарядное устройство и маркировка |
|
ГОСТ Р ИСО 16107 |
Смещение относительно заданной изготовителем частоты отбора проб, вариация в частоте отбора проб (например, из-за плохого допуска на размеры пробоотборника), влияния ветра, температуры, влажности, самой целевой концентрации и диффузионных потерь от пробоотборника |
|
ГОСТ Р ЕН 838 |
Предел количественного определения методики измерений, аналитическое извлечение, номинальная скорость накопления, скорость воздуха/ориентация пробоотборника, испытание на утечку пробоотборника, срок годности и маркировка |
|
ГОСТ Р ЕН 13205 |
Изменчивость пробы, безопасность конструкции насоса и электробезопасность, а также оценка пробоотборника |
6 Метод испытаний
6.1 Оценивают расширенную неопределенность результатов, полученных с использованием методики измерений, при проведении ее испытаний, предписанных соответствующими стандартами, специфичными для типа методики процедуры или соответствующего устройства, как указано в разделе 2.
Проводят измерения при концентрации вредного вещества, близкой к нижнему и верхнему значениям диапазона измерений, приведенных в таблице 1, и по крайней мере при одной промежуточной концентрации.
Готовят как минимум шесть параллельных проб для каждого набора испытаний и анализируют пробы в условиях повторяемости.
Вычисляют расширенную неопределенность, выраженную в процентах, по следующей процедуре (см. [12], [13], [14] и ГОСТ 34100.3):
a) указывают измеряемую величину;
b) выявляют все возможные источники составляющих неопределенности;
c) количественно оценивают случайную
и систематическую
составляющие неопределенности отбора проб;
________________
В ряде публикаций составляющие неопределенности разделяют на случайные и систематические, связывая их с погрешностями, возникающими соответственно из случайных и реальных систематических эффектов. Такая классификация составляющих неопределенности может привести к неоднозначности толкования при ее практическом применении. Например, случайная составляющая неопределенности в одном измерении может стать систематической составляющей в другом измерении, в котором результат 1-го измерения использован в качестве входных данных. При классификации методов оценивания составляющих неопределенности, а не самих составляющих, такая неоднозначность устраняется. В то же время это не препятствует объединению отдельных составляющих, оценка которых проведена двумя разными методами (оценки по типу A и типу B), в группы для конкретных целей (см.
ГОСТ 34100.3-2017**
, п.3.4.3).
d) количественно оценивают случайную
и систематическую
составляющие неопределенности анализа (аналитической стадии методики);
e) вычисляют суммарную случайную составляющую стандартной неопределенности
согласно формуле (1) и суммарную систематическую составляющую стандартной неопределенности
согласно формуле (2):
, (1)
; (2)
f) вычисляют суммарную стандартную неопределенность
согласно формуле
; (3)
g) вычисляют расширенную неопределенность
, используя коэффициент охвата
, значение которого, как правило, составляет от 2 до 3 согласно формуле
. (4)
Примечания
1 В качестве альтернативы случайные и систематические составляющие неопределенности отбора проб и случайные и систематические составляющие неопределенности анализа могут быть объединены в другом порядке для расчета суммарной стандартной неопределенности.
2 Приложение B содержит информацию о различных составляющих неопределенности отбора проб и анализа.
3 Подробные методы расчета приведены в ГОСТ Р ЕН 838 и ГОСТ Р ЕН 13205.
4 Расширенная неопределенность связана с концепцией точности измерений, используемой в некоторых сферах как диапазон, содержащий 95% результатов измерений определенной величины. Например, оценки точности, опубликованные в руководстве по аналитическим методам NIOSH [15], предоставляют значения расширенной неопределенности, скорректированные, если необходимо, для учета всех соответствующих источников изменчивости. Метод, соответствующий критерию точности NIOSH (с расширенной неопределенностью, намного меньшей, чем 25%), вероятно, также будет отвечать требованиям к эффективности (5.4.5), установленным настоящим стандартом (см. [16], [17], [18], [19]).
5 Для оценки показателей неопределенности измерений на аналитической стадии методики измерений или в целом для методики измерений, не предусматривающей накопления химического вещества в пробоотборнике (например, отбор проб воздух рабочей зоны в газовые пипетки или мешки), может применен [20], алгоритмы которого позволяют получать статистически достоверные оценки показателей неопределенности.
6.2 При необходимости проводят дальнейшие испытания для изучения влияния мешающих веществ и условий окружающей среды, например: скорости, направления ветра или ориентации пробоотборника.
6.3 Для методики измерений, состоящей из нескольких этапов (подготовка оборудования, отбор проб, транспортирование и хранение пробы, анализ), испытание каждого ее этапа может быть проведено отдельно вместо испытания методики в целом. В этом случае вычисляют относительную расширенную неопределенность результатов, полученных по методике измерений, путем соответствующего объединения неопределенностей всех независимых этапов.
Примечание — Для некоторых химических веществ характеристику эффективности одного этапа или более следует определять другими средствами без использования в испытании самого химического вещества. Подробное описание таких случаев приведено в соответствующих стандартах.
7 Отчет о валидации
Отчет о валидации следует составлять для каждой методики измерений, прошедшей испытания, с указанием, по крайней мере, условий проведения испытаний, полученных результатов и вывода о степени соответствия методики измерений требованиям настоящего или другого соответствующего стандарта.
Приложение A
(обязательное)
Структура описания метода
Пример структуры описания метода приведен ниже. Применение положений пунктов или подпунктов определяют в каждом конкретном случае в индивидуальном порядке. В данном описании представлена вся необходимая информация для выполнения методики измерений, включая информацию о достижимой расширенной неопределенности, диапазоне измерений, времени усреднения, мешающих веществах, а также о условиях окружающей среды или других условиях, которые могут повлиять на показатели эффективности методики измерений.
При необходимости, должны быть приведены следующие основные положения:
— введение;
— наименование;
— требования безопасности;
— область применения;
— нормативные ссылки;
— термины и определения;
— принцип;
— требования, включая заданный диапазон измерений, температурную стабильность при транспортировании, мешающие вещества, влияние окружающей среды, опасность взрыва, механическую прочность и долговечность, инструкции по применению и т.д.;
— условия испытаний, включая реакции, реактивы и материалы, аппаратуру и оборудование, генерацию тестового газа и заданные условия испытаний;
— методы испытаний, включая методики испытаний, механическую прочность и долговечность, опасность взрыва, отбор проб, транспортирование и хранение, аналитическую процедуру и расчет концентрации;
— обеспечение качества и контроль качества;
— особые случаи (например, наличие мешающих веществ);
— неопределенность измерений;
— отчет об испытании;
— приложения.
Описание методик измерений, применяемых в сфере государственного регулирования обеспечения единства измерений, должно соответствовать требованиям ГОСТ Р 8.563**.
Приложение B
(справочное)
Расчет неопределенности измерений
B.1 Общие положения
Первым шагом в оценке неопределенности измерений в соответствии с ГОСТ 34100.3 является определение возможных источников неопределенности и выявление отдельных случайных и систематических составляющих неопределенности (см. [12], [13], [14] и ГОСТ 34100.3). Для оценки составляющих неопределенности может быть полезно построение диаграммы причинно-следственных связей.
Методики измерений химических веществ включают два основных этапа: отбор проб и анализ. Ниже приведен типичный, но неисключительный перечень случайных и систематических составляющих неопределенности:
a) неопределенность отбора проб, связанная:
1) с объемом отобранного воздуха или накопленной массой (см. B.2),
2) с эффективностью отбора проб (см. B.3),
3) с хранением, обработкой и/или транспортированием проб (см. B.4);
b) неопределенность анализа газов и паров (см. ГОСТ Р ЕН 838), связанная:
1) с извлечением метода (включает несколько факторов, таких как аналитическое извлечение, смещение метода, референтную концентрацию, влияние температуры, влияние относительной влажности и т.д.) (см. B.5),
2) с вариабельностью метода (включает такие факторы, как данные о прецизионности метода, полученные из результатов анализа повторных проб, концентрация определяемого вещества в растворах для градуировки, градуировочная функция, разбавления растворов проб, если применимо, дрейф отклика прибора и т.д.) (см. B.7);
c) неопределенность анализа частиц в воздухе (см. [3]) и смесей частиц воздуха и пара, связанная:
1) с аналитическим извлечением (см. B.6),
2) с аналитической вариабельностью (см. B.8),
3) с вычитанием холостой пробы (см. B.9).
Каждую из этих составляющих неопределенности оценивают или рассчитывают, а затем объединяют для получения оценки неопределенности метода измерений в целом, как описано в разделе 6.
При прямоугольном распределении вероятности или треугольном распределении вероятности диапазон ±A следует преобразовать в систематическую неопределенность, равную A/
или A/
соответственно.
B.2 Неопределенность, связанная с объемом отобранной пробы воздуха или накопленной массой
B.2.1 Отбор проб прокачкой
B.2.1.1 Источники неопределенности
При отборе пробы прокачкой для соответствующего объема воздуха имеются следующие источники неопределенности: измерение объемного расхода (см. B.2.1.2), стабильность прокачиваемого потока (см. B.2.1.3) и время отбора пробы (см. B.2.1.4).
B.2.1.2 Измерение объемного расхода
Измерения объемного расхода могут быть выполнены с использованием ряда различных устройств, например: ротаметров, массовых расходомеров, пузырьковых расходомеров или расходомеров с сухим поршнем. Ошибка измерений объемного расхода возникает из трех источников: калибровка расходомера (систематическая составляющая), показания расходомера (случайная составляющая) и, при необходимости, коррекция показаний расхода в соответствии с давлением и температурой окружающей среды.
Неопределенность калибровки расхода
должна быть оценена на основе данных, приведенных в сертификате испытания расходомера.
Неопределенность показаний скорости потока
следует принимать как выборочный коэффициент вариации, полученный по наблюдениям в условиях повторяемости.
Примеры неопределенности измерений расхода для разных типов расходомеров приведены в таблице B.1.
Если скорость потока измеряется несколько раз, а не только в начале отбора проб, неопределенность показаний скорости потока уменьшается в 1/
раз, где
— количество измерений скорости потока.
Таблица B.1 — Неопределенность измерений расхода для разных типов расходомеров (пример данных)
|
Тип расходомера |
Шкала, % |
Неопределен- ность калибровки расхода , % |
Неопределен- ность показаний расхода , % |
|
|
Ротаметр, длина 30 |
100 |
1,6 |
0,23 |
|
|
см
|
50 |
2,0 |
0,45 |
|
|
10 |
5,2 |
2,3 |
||
|
Тип расходомера |
Диапазон измерений расходомера, дм /мин |
Измеренный расход потока, дм /мин |
Неопределен- ность калибровки расхода , % |
Неопределен- ность показаний расхода , % |
|
Массовый расходомер |
От 0,1 до 15 |
2,0 |
0,61 |
2,0 |
|
Пузырьковый расходомер |
От 0 до 0,25 |
0,12 |
0,4 |
0,35 |
|
От 0,2 до 6 |
2,0 |
0,12 |
0,1 |
|
|
От 2 до 30 |
3,0 |
0,06 |
0,22 |
|
|
Сухой поршневой расходомер |
От 0,5 до 5 |
2,0 |
0,59 |
0,26 |
|
От 0,5 до 25 |
3,0 |
0,41 |
0,07 |
|
|
Неопределенность калибровки расхода предполагает прямоугольное распределение вероятностей и рассчитывается с использованием данных из сертификата калибровки расходомера. Неопределенность показаний расхода основана на 10 измерениях. Неопределенность показаний расхода аналогового расходомера зависит от разрешения шкалы прибора. |
B.2.1.3 Стабильность потока
Насосы для индивидуального отбора проб воздуха, как правило, являются саморегулирующимися и поддерживают установленный расход независимо от изменения обратного давления. В ГОСТ Р ИСО 13137 установлено, что скорость потока должна быть в пределах ±5% от указанного значения в течение всего периода отбора проб. При прямоугольном распределении вероятности максимально допустимое значение систематической составляющей неопределенности стабильности прокачиваемого потока составляет 5/
%.
Оценка фактических значений стабильности прокачиваемого потока может быть проведена по значению, указанному производителем, или по результатам испытаний, приведенных в ГОСТ Р ИСО 13137, и составлять менее чем 5%. При прямоугольном распределении вероятности значение систематической составляющей неопределенности стабильности прокачиваемого потока и
может быть рассчитано по формуле
, (В.1)
где
— разница между средним показанием расхода при минимальном и максимальном обратном давлении, %.
В.2.1.4 Время отбора проб
Время отбора проб может быть очень точно измерено с помощью радиоуправляемых часов, кварцевых часов или секундомера, которые имеют калибровку, прослеживаемую к национальному эталону времени. Основным источником неопределенности при измерении времени отбора проб является точность, с которой производится считывание, т.е. с точностью до минуты или секунды.
Если показание считывается с точностью до секунды, систематическая составляющая неопределенности предельна мала как для долгосрочных, так и для краткосрочных измерений и может быть незначительной. Если считывание производится до ближайшей минуты, систематическая составляющая предельна мала для долгосрочных измерений (например, более 2 ч) и может быть проигнорирована, но для краткосрочных измерений ее необходимо учитывать.
Например, если время фиксируют с точностью до ближайшей минуты, коэффициент вариации составляет 2,7% для времени отбора проб 15 мин (суммируя максимальные отклонения 0,5 мин в начале и конце периода отбора и деля на время отбора пробы и
, предполагая треугольное распределение вероятности).
В случае отбора проб прокачкой согласно ГОСТ Р ИСО 13137 указанное время не должно отклоняться более чем на ±0,5% для таймера, прошедшего калибровку. При прямоугольном распределении вероятности максимально допустимое значение систематической составляющей неопределенности составляет 0,5
=0,29%.
B.2.2 Диффузионный отбор проб
B.2.2.1 Источники неопределенности
Для диффузионного отбора проб накопление массы имеет следующие источники неопределенности: скорость накопления (см. В.2.2.2) и время отбора (см. В.2.2.3).
B.2.2.2 Скорость накопления
Случайные и систематические составляющие неопределенности скорости накопления следует оценивать по повторным пробам, полученным из испытательной атмосферы, как описано в ГОСТ Р ЕН 838.
B.2.2.3 Время отбора проб
Информация относительно данного параметра приведена в B.2.1.4.
B.3 Неопределенность, связанная с эффективностью отбора проб
B.3.1 Методы отбора проб для газов и паров прокачкой
Отбор проб газов и паров прокачкой может зависеть от давления, влажности и температуры отбираемого воздуха, концентрации химических веществ в отбираемом воздухе и объемного расхода. Данные факторы могут повлиять на производительность и эффективность процесса отбора проб. Неопределенность, связанная с этими эффектами, включена в составляющую неопределенности извлечения метода. Однако для прокачиваемых пробоотборников объем пробы держится значительно ниже экспериментально установленного объема проскока, и в таком случае эффективность отбора проб предполагается равной 100%, и неопределенность эффективности отбора проб не требуется принимать во внимание.
B.3.2 Методы диффузионного отбора проб для газов и паров
Для диффузионного отбора проб эффективность отбора имеет следующие источники неопределенности — обратная диффузия и время воздействия.
Обратная диффузия может происходить, если в течение периода отбора проб наблюдается значительная вариация концентрации химического вещества в воздухе. На нее влияют характеристики сорбента и химического вещества, давление, влажность и температура отбираемого воздуха и масса отобранного химического вещества. (Последняя является функцией концентрации химического вещества в отобранном воздухе и времени отбора пробы.) Оценка систематической составляющей неопределенности из-за обратной диффузии может быть проведена по разнице средних значений результатов в двух наборах повторных проб. Пробы получают путем воздействия на диффузионные пробоотборники в течение короткого периода времени высокой концентрацией химического вещества, впоследствии один из пробоотборников подвергается воздействию чистого воздуха в течение длительного периода времени, как описано в ГОСТ Р ИСО 16107 и ГОСТ Р ЕН 838.
Систематическая составляющая неопределенности, связанная со временем воздействия, может быть оценена путем анализа повторяющихся проб, отобранных в испытательной среде, согласно ГОСТ Р ИСО 16107 и ГОСТ Р ЕН 838.
B.3.3 Методы отбора проб аэрозолей
B.3.3.1 Общие положения
Для методов отбора аэрозольных проб эффективность отбора проб имеет следующие источники неопределенности: близость соответствия требуемым положениям по отбору проб и эффективность подложки для накопления.
B.3.3.2 Близость соответствия требуемым положениям по отбору проб
Каждая стадия накопления на пробоотборнике для отбора частиц в воздухе должна следовать положениям об отборе проб для одной из фракций, связанных со здоровьем, как описано в ГОСТ Р ИСО 7708. Методы отбора проб аэрозоля имеют случайные и систематические составляющие неопределенности в зависимости от того, в какой степени используемые пробоотборники соответствуют требуемым положениям по отбору проб.
В [8] и [9] описаны методы испытаний для определения правильности отбора пробоотборником требуемую(ые) аэрозольную(ые) фракцию(и). В соответствии с методом согласно [8] проводят расчет определения кривой средней эффективности отбора проб исходя из отдельных значений эффективности отбора проб испытуемого пробоотборника в зависимости от аэродинамического диаметра частиц. Эффективность отбора проб рассчитывают на основе определенной концентрации аэрозоля в пробах, отобранных пробоотборником при испытании, деленное на общую концентрацию аэрозоля в окружающей среде, оцененное по значениям изокинетического пробоотборника по крайней мере для девяти аэродинамических размеров частиц. В методе, приведенном в [9], сравнивают концентрацию, измеренную потенциальным пробоотборником, и концентрацию, измеренную валидированным (референтным) пробоотборником по крайней мере для трех испытательных аэрозолей с наиболее различающимися распределениями частиц по размерам. При этом кривую эффективности отбора проб определить невозможно.
B.3.3.3 Составляющие неопределенности для аэрозольных пробоотборников — оценки для общего использования
Данные по эффективности отбора проб экспериментально определены и опубликованы для различных типов пробоотборников для улавливания вдыхаемой, торакальной и респирабельной фракций. Исходя из этих данных, [8] предоставляет диапазоны для различных составляющих неопределенности для подобных пробоотборников. Однако в настоящее время суммарная стандартная неопределенность для конкретных пробоотборников не была рассчитана в соответствии с требованиями настоящего стандарта.
Примечание — Вместо вычисления суммарной стандартной неопределенности для конкретного пробоотборника можно временно использовать информативные оценки составляющих неопределенности, приведенные в ГОСТ Р ИСО 21748.
B.3.3.4 Эффективность подложки для накопления
B.3.3.4.1 Фильтрующие материалы
Фильтрующие материалы должны быть выбраны таким образом, чтобы иметь высокую эффективность накопления для необходимого диапазона размеров частиц, и в этом случае неопределенность, связанная с эффективностью накопления, незначительна.
B.3.3.4.2 Вспененные материалы
Когда в качестве подложки для накопления используют вспененный материал, эффективность отбора проб и эффективность накопления взаимосвязаны, и необходимость добавлять составляющие неопределенности отсутствуют.
B.4 Неопределенность, связанная с хранением и транспортированием проб
B.4.1 Хранение проб
Систематическая составляющая неопределенности, связанная с хранением проб, может быть оценена путем анализа проб, собранных из испытательной атмосферы или приготовленных путем добавления в средство для отбора проб необходимого химического вещества. Ее можно рассчитать по разнице
между средними результатами повторных проб, проанализированных непосредственно после отбора проб/добавления химического вещества, и повторных проб, проанализированных после максимального периода хранения, указанного в методе испытаний. При прямоугольном распределении вероятности разность
можно разделить на
. Испытания на хранение описаны в ГОСТ Р ЕН 838.
B.4.2 Транспортирование проб
B.4.2.1 Пробы газа и пара
Когда пробы транспортируют соответствующим образом согласно методики измерений, составляющая неопределенности, связанная с транспортированием, может быть незначительной.
B.4.2.2 Пробы аэрозоля
При транспортировании подложек с накопленным аэрозолем, как правило, возникает неопределенность, связанная с миграцией пыли из подложки для накопления в ее держатель, или наоборот. Загрязнение пробоотборной кассеты может быть основным источником неопределенности, так как ее масса контролируется. Испытание целостности при транспортировании описано в ГОСТ Р ИСО 15767 и [10]. Требуется, чтобы относительные потери пыли при транспортировании были менее 5% в соответствии с ГОСТ Р ИСО 15767. Все относительные изменения массы должны быть менее или равны 5% для загрузок проб, соответствующих концентрации в диапазоне от 0,5 до 2 ПЗПВ, и 15% для загрузок проб, соответствующих концентрации в диапазоне от 0,1 до 0,5 по ГОСТ Р ИСО 15767 или [10]. Верхний предел загрузки подложки для накопления может быть определен согласно [10]. Систематическую составляющую неопределенности определяют исходя из критерия приемки для верхнего предела загрузки пробы.
B.5 Неопределенность, связанная с извлечением метода для газов и паров
На извлечение метода влияет несколько факторов. Данные факторы включают аналитическое извлечение, смещение метода, референтную концентрацию, влияние влажности и влияние температуры. Если коррекцию на аналитическое извлечение не применяют к результатам, то рассматривают это как составляющую неопределенности. Исследование такого влияния проводят с использованием испытательной атмосферы по ГОСТ Р ЕН 838 и [1]. Экспериментальные данные, собранные в результате проведения испытаний, предоставляют информацию о факторах, вызывающих вариацию и смещение (относительно референтного значения), которые возникают при рутинном применении установленной методики измерений (такие как концентрация, температура и влажность). Полученные данные могут быть использованы для оценки неопределенности метода в целом. Методика измерений для газов и паров, как правило, предписывает коррекцию результатов на аналитическое извлечение, в связи с чем оценку извлечения метода осуществляют по результатам анализа проб, отобранных из испытательной атмосферы, с корректировкой на аналитическое извлечение.
B.6 Неопределенность, связанная с аналитическим извлечением частиц в воздухе и смесей частиц в воздухе и пара
Смещение обычно устраняют при разработке аналитического метода, однако это не всегда возможно. В соответствии с ГОСТ 34100.3 результаты измерений должны быть скорректированы с учетом смещения, если оно является значительным. Однако это часто практически неосуществимо, например в методиках измерений металлов и металлоидов в пробах воздуха рабочей зоны, поскольку аналитическое смещение может варьироваться в зависимости от матрицы пробы. Следовательно, аналитическое смещение должно быть оценено и рассмотрено как составляющая неопределенности.
Согласно [3] оценка систематической составляющей неопределенности аналитического смещения может быть осуществлена с использованием:
— результатов анализа сертифицированных стандартных образцов и/или чистых соединений;
— результатов межлабораторных сличений;
— результатов испытаний по извлечению, проведенных с добавлением в лабораторные холостые пробы;
— приемлемого диапазона смещения.
Систематическая составляющая неопределенности также может быть принята равной нулю для методик, включающих эмпирическую пробоподготовку (например, для методики определения тех растворимых металлов и металлоидов, для которых использование, отдельных аналитических условий по определению приводит к правильному аналитическому результату без вклада неопределенности аналитического извлечения).
B.7 Неопределенность, связанная с вариабельностью метода для газов и паров
Оценка неопределенности, связанной с вариабельностью метода, может быть осуществлена исходя из данных прецизионности метода, полученных на основе результатов повторных проб, отобранных из испытательной атмосферы и используемых согласно ГОСТ Р ЕН 838. Отдельные оценки неопределенности должны быть сделаны для любых источников систематической ошибки в том случае, если систематические ошибки не могут быть исправлены. Примеры включают систематическую составляющую неопределенности, связанную с концентрацией растворов для построения градуировочной кривой, с градуировочной функцией, разбавлением растворов проб и дрейфом отклика прибора.
Неопределенность, связанная с аналитической вариабельностью, включена в вариабельность метода.
Независимые оценки неопределенности, связанные с аналитической вариабельностью, могут быть сделаны исходя из данных аналитической прецизионности, полученных либо в условиях повторяемости, либо в условиях воспроизводимости. В обоих случаях необходимо проводить отдельные оценки неопределенности для любых источников систематической ошибки, где это применимо (например, для систематической составляющей неопределенности, связанной с концентрацией растворов для построения градуировочной кривой, с градуировочной функцией, разбавлением растворов проб и дрейфом отклика прибора). Когда аналитическую прецизионность определяют на основе данных внутрилабораторной прецизионности (например, данных контроля качества), то включают большинство случайных и систематических составляющих неопределенности аналитической вариабельности (см. ГОСТ Р ИСО 21748). При использовании данных внутрилабораторной прецизионности значения, полученные для аналитической прецизионности, могут быть выше, чем при применении данных повторяемости, потому что в этом случае включена прецизионность между разными днями.
B.8 Неопределенность, связанная с аналитической вариабельностью для частиц в воздухе и смеси частиц в воздухе и пара
Оценка неопределенности, связанной с аналитической вариабельностью, может быть осуществлена исходя из данных аналитической прецизионности, полученных либо в условиях повторяемости, либо в условиях воспроизводимости, согласно [3]. В обоих случаях отдельные оценки неопределенности должны быть сделаны для любых источников систематических ошибок, где это применимо (например, для систематической составляющей неопределенности, связанной с концентрацией растворов для построения градуировочной кривой, с градуировочной функцией, разбавлением раствора пробы и дрейфом отклика прибора). Когда аналитическую прецизионность определяют по данным лабораторной прецизионности (например, по данным контроля качества), включают большинство случайных и систематических составляющих неопределенности аналитической изменчивости (см. ГОСТ Р ИСО 21748).
B.9 Холостые подложки
Случайная составляющая неопределенности, связанная с холостыми подложками, должна быть включена в бюджет неопределенности, если результаты измерений проб корректируют с учетом холостой подложки, как описано в ГОСТ Р ИСО 15767, или должна быть включена систематическая составляющая неопределенности, если корректировка по холостой не выполнена.
B.10 Общее у равнение для суммарной неопределенности составляющих
Для расчета случайных и систематических составляющих неопределенности отбора проб и анализа отдельные составляющие суммируются в соответствии с формулами (B.2)-(B.5) [см. также 6.1 (примечание 1) и 6.3 (примечание)]
, (B.2)
, (В.3)
, (В.4)
, (В.5)
где
,
,
и
— случайная составляющая неопределенности отбора проб, систематическая составляющая неопределенности отбора проб, случайная составляющая неопределенности анализа и систематическая составляющая неопределенности анализа, соответственно;
,
,
и
— соответствующие отдельные составляющие неопределенности;
,
,
и
— соответствующие номера отдельных составляющих неопределенности.
Приложение ДА
(справочное)
Сведения о соответствии ссылочных национальных и межгосударственных стандартов международным и европейским стандартам, использованным в качестве ссылочных в примененном международном стандарте
Таблица ДА.1
|
Обозначение ссылочного национального, межгосударственного стандарта |
Степень соответствия |
Обозначение и наименование ссылочного международного, европейского стандарта |
|
ГОСТ Р ИСО 7708-2006 |
IDT |
ISO 7708 «Качество воздуха. Определение гранулометрического состава частиц при санитарно-гигиеническом контроле» |
|
ГОСТ Р ИСО 13137-2016 |
IDT |
ISO 13137 «Воздух рабочей зоны. Насосы для индивидуального отбора проб химических и биологических веществ. Требования и методы испытаний» |
|
ГОСТ Р ИСО 16107-2009 |
IDT |
ISO 16107 «Воздух рабочей зоны. Оценка характеристик диффузионных пробоотборников» |
|
ГОСТ Р ЕН 838-2010 |
IDT |
EN 838 «Воздух рабочей зоны. Диффузионные пробоотборники, используемые при определении содержания газов и паров. Требования и методы испытаний» |
|
ГОСТ Р ЕН 13205-2010 |
IDT |
EN 13205 «Воздух рабочей зоны. Оценка характеристик приборов для определения содержания твердых частиц» |
|
ГОСТ Р ИСО 15767-2012 |
IDT |
ISO 15767 «Воздух рабочей зоны. Контроль и оценка неопределенности взвешивания проб аэрозолей» |
|
ГОСТ 34100.3-2017 |
IDT |
ISO/IEC Guide 98-3:2008 «Неопределенность измерения. Часть 3. Руководство по выражению неопределенности измерения» |
|
ГОСТ Р ИСО 21748-2012 |
IDT |
ISO 21748 «Статистические методы. Руководство по использованию оценок повторяемости, воспроизводимости и правильности при оценке неопределенности измерений» |
|
Примечание — В настоящей таблице использовано следующее условное обозначение степени соответствия стандартов: — IDT — идентичные стандарты. |
Библиография
|
[1] |
ЕН 1076 Воздействие на рабочем месте. Методики измерений газов и паров с использованием насосных пробоотборников. Требования и методы испытаний |
|
[2] |
ИСО 17621 Воздух рабочей зоны. Газоопределители с колористической индикаторной трубкой для измерений разовых концентраций. Требования и методы испытаний |
|
[3] |
ЕН 13890 Воздействие на рабочем месте. Методики измерений металлов и металлоидов в частицах, находящихся в воздухе. Требования и методы испытаний |
|
[4] |
ЕН 13936 Воздействие на рабочем месте. Методики измерений химического вещества, присутствующего в виде смеси частиц и паров в воздухе. Требования и методы испытаний |
|
[5] |
ЕН 45544 Воздух рабочей зоны. Электрические аппараты, используемые для прямого обнаружения и прямого измерения концентрации токсичных газов и паров |
|
[6] |
РМГ 29-2013 Государственная система обеспечения единства измерений. Метрология. Основные термины и определения |
|
[7] |
ИСО 78-2 Химия. Макеты стандартов. Часть 2. Методы химического анализа |
|
[8] |
ЕН 13205-2, Воздействие внешних факторов на рабочее место. Оценка технических характеристик пробоотборников для измерения концентрации взвешенных в воздухе частиц. Часть 2. Лабораторная проверка рабочих характеристик на основе определения эффективности отбора проб |
|
[9] |
ЕН 13205-4, Воздействие внешних факторов на рабочее место. Оценка технических характеристик пробоотборников для измерения концентрации взвешенных в воздухе частиц. Часть 4. Лабораторная проверка рабочих характеристик на основе сравнения концентраций |
|
[10] |
ЕН 13205-6, Воздействие внешних факторов на рабочее место. Оценка технических характеристик пробоотборников для измерения концентрации взвешенных в воздухе частиц. Часть 6. Испытания по переносу аэрозолей и обращение с ними |
|
[11] |
ГН 2.2.5.3532-18 «Предельно допустимые концентрации (ПДК) вредных веществ в воздухе рабочей зоны»
|
________________
Следует использовать вместо базы данных «Database: GESTIS — International limit values for chemical agents, http://www.dguv.de/ifa/GESTIS/GESTIS-lnternationale-Grenzwerte-
-chemische-Substanzen-limit-values-for-chemical-agents/index-2.jsp».
|
[12] |
Nordtest, Handbook for calculation of measurement uncertainty in environmental laboratories, Version 3.1 (May 2012), Nordtest Technical Report, NT TR 537-Edition 3.1, November 2012, http://www.nordtest.info/index.php/technical-reports/item/handbook-for- calculation-of-measurement-uncertainty-in-environmental-laboratories-nt-tr-537-edition-3.html |
|
[13] |
European Co-operation for Accreditation, Expression of Uncertainty of Measurement in calibration, EA-4/02, www.european-accreditation.org |
|
[14] |
EuraChem, Quantifying Uncertainty in Analytical Measurement, www.measurementuncertainty.org |
|
[15] |
NIOSH Manual of Analytical Methods. DHHS (NIOSH) Publications No. 94-113, No. 96-135, No. 98-119, and No. 2003-154, NIOSH, Cincinnati, OH, 1994. Available at www.cdc.gov/niosh/nmam [Accessed March 25 2015] |
|
[16] |
Bartley D.L. Definition and assessment of sampling and analytical accuracy. Ann. Occup. Hyg. 2001, 45 pp.357-364 |
|
[17] |
Bartley D.L., Shulman S.A., Schlect P.C. Measured Uncertainty and NIOSH Method Accuracy Range. In: NIOSH Manual of Analytical Methods, (Ashley K. ed.). NIOSH, Cincinnati, Fifth Edition, 2004, pp.208-228. |
|
[18] |
Bartley D.L., & G.M. Measurement uncertainty. J. Occup. Environ. Hyg. 2008, 52 pp.413-417 |
|
[19] |
Bartley D.L. Reconciling traditional accuracy assessment with the ISO Guide to the Expression of Uncertainty in Measurement (ISO/GUM). J. Occup. Environ. Hyg. 2004, 1 pp.D37-D41 |
|
[20] |
РМГ 61-2070 Государственная система обеспечения единства измерений. Показатели точности, правильности и прецизионности методик количественного химического анализа. Методы оценки |
|
УДК 504.3:006.354 |
ОКС 13.040.30 |
|
Ключевые слова: воздух, рабочая зона, неопределенность, химические вещества, методика измерений, отбор проб, анализ |
| Найти: | |
| Где: | |
| Тип документа: | |
| Отображать: | |
| Упорядочить: |
Скачать 4945-88 Методические указания по определению вредных веществ в сварочном аэрозоле (твердая фаза и газы)
Дата актуализации: 17.06.2011
4945-88
Методические указания по определению вредных веществ в сварочном аэрозоле (твердая фаза и газы)
| Статус: | действует |
| Обозначение: | 4945-88 |
| Название рус.: | Методические указания по определению вредных веществ в сварочном аэрозоле (твердая фаза и газы) |
| Дата актуализации текста: | 01.01.2009 |
| Дата добавления в базу: | 29.04.2009 |
| Дата введения: | 22.12.1988 |
| Разработан в: | Минздрав СССР |
| Утверждён в: | Главный государственный санитарный врач СССР (22.12.1988) |
| Опубликован в: | МП «Рарог» № 1992 |
| Область и условия применения: | Настоящие методические указания предназначены для санитарных лабораторий промышленных предприятий и учреждений санитарно-эпидемиологической службы, осуществляющих контроль за содержанием вредных веществ в воздухе рабочей зоны, а также организаций и специалистов, проводящих работы по гигиенической оценке сварочных материалов и способов сварки, наплавки и термической резки металлов, являющихся источником выделения сварочных аэрозолей (СА), с целью проведения оздоровительных мероприятий и оценки их эффективности. |
| Заменяет собой: |
|
| Оглавление: | 1 ОБЩАЯ ХАРАКТЕРИСТИКА СВАРОЧНЫХ АЭРОЗОЛЕЙ 2. ОСНОВНЫЕ ТРЕБОВАНИЯ К ОТБОРУ ПРОБ ВОЗДУХА 3. МЕТОДЫ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ 3.1. ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ РАЗДЕЛЬНОЕ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ЖЕЛЕЗА, НИКЕЛЯ, МАРГАНЦА, ТИТАНА И ОКСИДОВ ХРОМА (III И VI) ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ МЕДИ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ЦИНКА И ОКСИДА ЦИНКА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ МОЛИБДЕНА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ КОБАЛЬТА И ОКСИДА КОБАЛЬТА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДОВ ВАНАДИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИЙ ВОЛЬФРАМА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ЦИРКОНИЯ И ОКСИДА ЦИРКОНИЯ (IV) ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ АЛЮМИНИЯ И ОКСИДА АЛЮМИНИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ МАГНИЯ И ОКСИДА МАГНИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ СВИНЦА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ БОРНОЙ КИСЛОТЫ И БОРНОГО АНГИДРИДА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ДИОКСИДА КРЕМНИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИЙ ФТОРИСТОГО ВОДОРОДА И СОЛЕЙ ФТОРИСТОВОДОРОДНОЙ КИСЛОТЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОЗОНА МЕТОД 1 МЕТОД 2 ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДОВ АЗОТА (II) И (IV) 3.2. ПОЛЯРОГРАФИЧЕСКИЕ МЕТОДЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИЙ МЕДИ, НИКЕЛЯ, КАДМИЯ, ЦИНКА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИЙ МЕДИ, НИКЕЛЯ И КОБАЛЬТА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ЖЕЛЕЗА, ТИТАНА, МОЛИБДЕНА, ОКСИДОВ ХРОМА (III И VI) И ВАНАДИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ЖЕЛЕЗА И МАРГАНЦА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ВОЛЬФРАМА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ МОЛИБДЕНА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ТИТАНА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ СВИНЦА, ОЛОВА, КАДМИЯ И МЕДИ 3.3. ИОНОМЕТРИЧЕСКИЕ МЕТОДЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИЙ БОРНОЙ КИСЛОТЫ И БОРНОГО АНГИДРИДА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ФТОРИСТОГО ВОДОРОДА И СОЛЕЙ ФТОРИСТОВОДОРОДНОЙ КИСЛОТЫ 3.4. АТОМНО-АБСОРБЦИОННЫЕ МЕТОДЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ КОБАЛЬТА, НИКЕЛЯ, МЕДИ, ЦИНКА, КАДМИЯ, СВИНЦА, ЖЕЛЕЗА, МАРГАНЦА, МОЛИБДЕНА, ОЛОВА, ВОЛЬФРАМА, ОКСИДА ВАНАДИЯ И ОКСИДОВ ХРОМА ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДА АЛЮМИНИЯ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДА КАЛЬЦИЯ 3.5. ГАЗОХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДА УГЛЕРОДА* 3.6. ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ ИЗМЕРЕНИЕ КОНЦЕНТРАЦИИ ОКСИДА КАЛЬЦИЯ ПРИЛОЖЕНИЯ Приложение 1 ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ВРЕДНЫХ ВЕЩЕСТВ, ВЫДЕЛЯЮЩИХСЯ В ВОЗДУХ ПРИ ПРОВЕДЕНИИ ДУГОВЫХ И ПЛАЗМЕННЫХ ПРОЦЕССОВ Приложение 2 ПРЕДЕЛЬНО-ДОПУСТИМЫЕ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ, ВЫДЕЛЯЮЩИХСЯ В ВОЗДУХ В ВИДЕ ТВЕРДОЙ И ГАЗОВОЙ СОСТАВЛЯЮЩИХ СВАРОЧНЫХ АЭРОЗОЛЕЙ Приложение 3 ПРАВИЛА ТЕХНИКИ БЕЗОПАСНОСТИ ПРИ РАБОТЕ В ЛАБОРАТОРИИ С ВРЕДНЫМИ ВЕЩЕСТВАМИ И АППАРАТУРОЙ Приложение 4 РАСЧЕТ КОНЦЕНТРАЦИИ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ 4.1. Приведение объема отобранного воздуха к стандартным условиям 4.2. Расчет результатов анализа с использованием градуировочных графиков 4.3. Расчет результатов анализа с использованием метода добавки Приложение 5 РАСЧЕТ ПОГРЕШНОСТИ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИЙ ВРЕДНЫХ ВЕЩЕСТВ В ВОЗДУХЕ Приложение 6 Примерный перечень основных элементов и соединений, выделяющихся в составе ТССА н ГССА при процессах сварки, наплавки, напыления и резки металлов, а также наиболее характерные и опасные вредные вещества (III III), по которым необходимо проводить контроль воздушной среды в зоне дыхания работающих и в воздухе рабочей зоны Приложение 7 |
| Расположен в: | Строительная документация Нормативные документы по надзору в области строительства Нормативные документы по санитарно-эпидемиологическому надзору |
Скачать 4945-88