Правила надлежащей клинической практики
(утв. приказом Министерства здравоохранения РФ от 1 апреля 2016 г. № 200н)
I. Общие положения
1. Настоящие Правила надлежащей клинической практики (далее — Правила) регулируют отношения по планированию, организации, проведению, мониторингу, аудиту, документированию клинических исследований лекарственных препаратов для медицинского применения (далее соответственно — клиническое исследование, лекарственный препарат) с участием человека в качестве субъекта клинического исследования, анализу и представлению результатов клинических исследований, обеспечивающие гарантию достоверности и точности полученных данных и представленных результатов, а также защиту прав, здоровья и конфиденциальности персональных данных участников клинического исследования.
2. Для целей настоящих Правил применяются термины и определения, используемые в Федеральном законе от 12 апреля 2010 г. № 61-ФЗ «Об обращении лекарственных средств» (далее — Федеральный закон «Об обращении лекарственных средств»).
3. Объектом клинического исследования является лекарственная форма фармакологически активного вещества или плацебо, изучаемые или используемые для контроля в клиническом исследовании, или зарегистрированный лекарственный препарат для медицинского применения в случае, если способ его применения отличается от утвержденного (в рамках процедуры государственной регистрации), а также при его использовании по новому показанию или для получения дополнительной информации по утвержденному показанию (далее — исследуемый лекарственный препарат). Исследуемые лекарственные препараты применяются в соответствии с утвержденным (в рамках процедуры получения разрешения на проведение клинического исследования) протоколом клинического исследования.
Обращение исследуемых лекарственных препаратов осуществляется в соответствии с правилами надлежащей производственной практики и правилами надлежащей практики хранения и перевозки лекарственных препаратов.
4. Клиническое исследование проводится в отношении физического лица (пациента или здорового добровольца), принимающего участие в клиническом исследовании в составе группы, получающей исследуемый лекарственный препарат, либо в составе контрольной группы (далее — участник клинического исследования).
Каждый участник клинического исследования после получения информации о клиническом исследовании и до начала проведения клинического исследования дает добровольное согласие на участие в таком клиническом исследовании посредством подписания информационного листка пациента.
Согласие на участие в клиническом исследовании может быть дано законным представителем участника клинического исследования.
5. Клиническое исследование лекарственного препарата проводится на основании разрешения на проведение клинического исследования, выданного Министерством здравоохранения Российской Федерации (далее — Министерство) по результатам экспертизы документов для получения разрешения на проведение клинического исследования и этической экспертизы, предусмотренных статьей 39 Федерального закона от 12 апреля 2010 г. № 61-ФЗ «Об обращении лекарственных средств».
6. Экспертиза документов, указанных в пункте 5 настоящих Правил, для получения разрешения на проведение клинического исследования лекарственного препарата проводится федеральным государственным бюджетным учреждением по проведению экспертизы лекарственных средств, этическая экспертиза осуществляется Советом по этике.
7. Клиническое исследование проводится в соответствии с протоколом клинического исследования (далее — протокол), который содержит, в том числе:
1) название протокола, идентификационный номер протокола и дату утверждения организатором исследования. Новые версии протокола имеют номер версии и дату, поправки к протоколу — номер поправки и дату;
2) наименование и адрес организации, осуществляющей организацию проведения клинического исследования и осуществляющей мониторинг (если они различные);
3) фамилию, имя, отчество (при наличии) и должность лица, уполномоченного от имени организации, осуществляющей организацию проведения клинического исследования, подписывать протокол и поправки к протоколу;
4) фамилию, имя, отчество (при наличии), должность, адрес и номер телефона медицинского эксперта данного клинического исследования, назначенного организацией, осуществляющей организацию проведения клинического исследования;
5) фамилию, имя, отчество (при наличии) и должность исследователя, отвечающего за проведение клинического исследования, а также адреса и номера телефонов клинических центров;
6) фамилию, имя, отчество (при наличии), должность, адрес и номер телефона врача, отвечающего за принятие решений медицинского характера (если данное лицо не является исследователем);
7) наименования и адреса клинических лабораторий и других медицинских и (или) технических служб и организаций, принимающих участие в клиническом исследовании;
обоснование клинического исследования, включающее:
а) название и описание исследуемых лекарственных препаратов (включая плацебо и активный контроль);
б) сводное изложение результатов доклинических исследований и клинических исследований (если ранее проводились) исследуемых лекарственных препаратов;
в) краткое описание известных и потенциальных рисков и пользы применения исследуемого лекарственного препарата для участников клинического исследования;
г) описание и обоснование способа введения, дозировки, режима дозирования и курса лечения;
д) указание на то, что клиническое исследование будет проводиться в соответствии с протоколом клинического исследования и настоящими Правилами;
е) описание исследуемой популяции;
ж) ссылки на литературные источники и данные, имеющие значение для клинического исследования и представляющие собой обоснование клинического исследования;
9) цели и задачи клинического исследования;
10) описание общего плана (дизайна) клинического исследования, включающего:
а) указание основных и дополнительных (при наличии) исследуемых параметров, которые будут оцениваться в ходе клинического исследования;
б) описание типа дизайна проводимого клинического исследования (двойное слепое, плацебо-контролируемое, параллельное и иные виды исследований) и графическую схему дизайна клинического исследования, процедур и этапов клинического исследования;
в) описание мер, направленных на минимизацию, исключение субъективности, в том числе рандомизации, слепого метода (маскировки) — метода, при применении которого одной или нескольким участвующим в клиническом исследовании сторонам неизвестно, какое лечение назначено участнику клинического исследования (простой слепой метод предусматривает неосведомленность участников клинического исследования о назначенном им виде лечения, двойной слепой метод подразумевает неосведомленность о назначенном им виде лечения участников клинического исследования, исследователей, и, в некоторых случаях, лиц, выполняющих статистическую обработку данных);
г) описание используемого в клиническом исследовании лечения, дозировок и схемы применения исследуемых лекарственных препаратов, описание лекарственной формы, упаковки и маркировки исследуемых лекарственных препаратов;
д) продолжительность участия пациентов или здоровых добровольцев в клиническом исследовании, описание последовательности и продолжительности всех периодов клинического исследования, в том числе периода последующего наблюдения, если таковой предусмотрен;
е) описание правил остановки частей клинического исследования и (или) клинического исследования в целом, критериев исключения для отдельных участников клинического исследования;
ж) описание процедуры учета исследуемых лекарственных препаратов, плацебо и препаратов сравнения;
з) описание способов хранения рандомизационных кодов и процедуры их раскрытия;
и) перечень данных, регистрируемых в индивидуальной регистрационной карте (без предварительной записи в письменном или электронном виде) и рассматриваемых в качестве первичных данных;
11) критерии включения участников в клиническое исследование;
12) критерии невключения участников в клиническое исследование;
13) критерии исключения участников из клинического исследования (основания прекращения применения исследуемого лекарственного препарата, исследуемого лечения);
14) сведения о всех используемых в клиническом исследовании лекарственных препаратах, включая их названия, дозировки, частоту приема, пути и способы введения, продолжительность лечения, периоды последующего наблюдения для каждой группы участников клинического исследования, сведения о разрешенных для применения участниками клинического исследования лекарственных препаратах (включая неотложную терапию) и запрещенных для применения до и (или) во время клинического исследования лекарственных препаратах, способах контроля за соблюдением участниками клинического исследования условий приема лекарственных препаратов;
15) перечень параметров эффективности исследуемого лекарственного препарата и методы, сроки проведения оценки, регистрации и анализа параметров эффективности лекарственного препарата;
16) перечень параметров безопасности исследуемого лекарственного препарата и методы и сроки проведения оценки, регистрации и анализа параметров безопасности исследуемого лекарственного препарата;
17) требования к отчетам, процедуре регистрации и сообщениям о нежелательных явлениях и интеркуррентных заболеваниях;
18) метод и продолжительность наблюдения за участниками клинического исследования после возникновения нежелательных явлений;
19) описание статистических методов клинического исследования, включающих, в том числе:
а) сроки проведения промежуточного анализа;
б) планируемое количество участников клинического исследования с обоснованием размера выборки;
в) применяемый уровень значимости клинического исследования;
г) критерии прекращения клинического исследования;
д) процедуры учета отсутствующих, не подлежащих анализу и сомнительных данных;
е) процедуры сообщения о любых отклонениях от первоначального статистического плана (все отклонения от первоначального статистического плана описываются и обосновываются в протоколе и (или) финальном отчете о клиническом исследовании);
ж) процедуры отбора участников клинического исследования для анализа (все рандомизированные участники клинического исследования, все участники клинического исследования, получившие хотя бы одну дозу исследуемого лекарственного препарата, все участники, соответствующие критериям отбора, участники клинического исследования, данные которых пригодны для оценки, иные категории участников);
20) описание действий по контролю качества и обеспечению качества;
21) описание этических аспектов клинического исследования;
22) описание работы с данными и ведению записей;
23) описание порядка финансирования клинического исследования и страхования участников клинического исследования;
24) указание на возможность публикации результатов клинического исследования.
8. Сводное изложение результатов доклинических и клинических исследований приводится в брошюре исследователя (далее — брошюра), содержащей, в том числе следующие сведения:
1) наименование организации-разработчика лекарственного препарата, номер клинического исследования, химическое, международное непатентованное, группировочное и торговое наименования лекарственного препарата, дату составления брошюры, номер версии брошюры, номер и дату предыдущей редакции брошюры, а также уведомление о конфиденциальности содержащейся в ней информации, указываемые на титульном листе брошюры;
2) краткое резюме о физических, химических, фармацевтических, фармакологических, токсикологических, фармакокинетических, метаболических и клинических свойствах исследуемого лекарственного препарата, соответствующих стадии клинической разработки исследуемого лекарственного препарата, обоснование для изучения исследуемого лекарственного препарата, его ожидаемые профилактические, терапевтические или диагностические показания, общий подход к оценке исследуемого лекарственного препарата;
3) описание физических, химических и фармацевтических свойств и лекарственной формы исследуемого лекарственного препарата, его компонентов (включая химические и (или) структурные формулы), обоснование (при необходимости) состава лекарственной формы, включая вспомогательные вещества;
4) результаты доклинических исследований фармакологических свойств, токсичности, фармакокинетики и метаболизма исследуемого лекарственного препарата, с описанием, в том числе:
а) использованных видов животных, их количество и пол в каждой группе;
б) единиц измерения дозы, кратности и пути введения, длительности курса введения;
в) системного распределения, продолжительности последующего наблюдения после окончания введения лекарственного препарата;
г) характера и частоты фармакологических или токсических эффектов, их выраженности или степени тяжести, дозозависимости эффектов, времени до наступления, обратимость, продолжительность эффектов;
д) доклинической фармакологии;
е) фармакокинетики, метаболизма и распределения исследуемого лекарственного средства в тканях животных всех видов, на которых проводились исследования, включая сведения о всасывании, местной и системной биодоступности исследуемого лекарственного средства и его метаболитов, а также их связь с данными фармакологических и токсикологических исследований на животных;
ж) токсикологии (описание токсических эффектов исследуемого лекарственного средства, выявленных в исследованиях на животных разных видов (если применимо), включая сведения о токсичности при однократном и многократном введении, канцерогенности, специальных исследованиях (местно-раздражающее и аллергизирующее действие), репродуктивной токсичности, генотоксичности (мутагенности).
5) описание эффектов применения исследуемого лекарственного препарата у человека, в том числе:
а) фармакокинетики (включая метаболизм, всасывание, связывание с белками плазмы, распределение и выведение) и биодоступности исследуемого лекарственного препарата, взаимодействия исследуемого лекарственного препарата (лекарственные взаимодействия, влияние приема пищи и иные взаимодействия), других имеющихся данных по фармакокинетике (результаты проведенных в рамках клинических испытаний фармакокинетических исследований на различных группах);
б) данных по безопасности, фармакодинамике, эффективности и дозозависимости эффектов исследуемого лекарственного препарата (и его метаболитов, если есть данные), полученных в ходе проведенных клинических исследований (с участием здоровых добровольцев и (или) пациентов) и интерпретация этих данных (по законченным клиническим исследованиям представляются сводные отчеты по эффективности и безопасности исследуемого лекарственного препарата, а также сводные таблицы нежелательных реакций по всем клиническим исследованиям, описываются значимые различия в характере и частоте нежелательных реакций);
в) пострегистрационного опыта применения исследуемого лекарственного препарата с указанием страны, в которых исследуемый лекарственный препарат был зарегистрирован и имеется в продаже;
6) инструкции для исследователя по диагностике и лечению возможных передозировок и нежелательных реакций при применении исследуемого лекарственного препарата, основанные на предыдущем клиническом опыте и фармакологических свойствах исследуемого лекарственного препарата, а также обобщенная информация по различным свойствам исследуемого лекарственного препарата.
9. Отчет о результатах клинического исследования (далее — отчет) составляется в письменной форме организацией, осуществляющей организацию проведения клинического исследования лекарственного препарата, на основании заключений медицинских организаций, проводивших это исследование и включает, в том числе:
1) титульную страницу, на которой указываются:
а) название отчета;
б) название лекарственного препарата;
в) изучаемое показание к применению лекарственного препарата;
г) краткое описание дизайна клинического исследования, наличие препарата сравнения, продолжительность клинического исследования, дозирование лекарственного препарата и контингент участников исследования;
д) наименование организации, осуществляющей организацию проведения клинического исследования;
е) идентификационный номер протокола клинического исследования;
ж) фазы клинического исследования;
з) даты начала и окончания клинического исследования;
и) дата составления отчета;
к) сведения об исследователе, руководителе медицинской организации, ответственном представителе организации, осуществляющей организацию проведения клинического исследования;
2) резюме — краткое описание проведенного клинического исследования с числовыми данными для иллюстрации результатов;
3) оглавление, включая перечень и расположение приложений, таблиц;
4) перечень сокращений и определение терминов, используемых в отчете;
5) названия медицинских организаций, в которых проводилось клиническое исследование, место нахождения, контактный телефон;
6) цели и задачи клинического исследования;
7) план проведенного клинического исследования, включающий, в том числе:
а) общий план (дизайн), план-описание клинического исследования и схематическое изображение этапов и процедур клинического исследования;
б) обоснование плана (дизайна) клинического исследования;
в) критерии выбора популяции;
г) назначенное лечение, идентификация исследуемых лекарственных препаратов, методы распределения участников клинического исследования по группам (рандомизация), дозы и время приема исследуемого лекарственных препаратов, предшествующая и сопутствующая терапия;
д) данные об эффективности и безопасности исследуемого лекарственного препарата (оценка и график определения показателей эффективности и безопасности);
е) способы обеспечения качества и достоверности полученных данных, включая данные аудита, результаты инспекций, если проводились;
ж) изменения, внесенные в протокол клиническое исследование;
9) сведения об отклонениях от протокола клинического исследования;
10) оценку эффективности лекарственного препарата, в том числе:
а) совокупность данных, подлежавших анализу при проведении клинического исследования;
б) демографические и (или) другие исходные данные;
в) сведения о соблюдении участниками клинического исследования схемы лечения;
г) результаты оценки эффективности: статистические, аналитические данные, выводы относительно эффективности;
11) сведения о безопасности лекарственного препарата, в том числе:
а) нежелательные реакции (краткое резюме о нежелательных реакциях, их анализ, списки нежелательных реакций, которые наблюдались у всех участников клинического исследования);
б) смерть и другие серьезные нежелательные явления;
в) оценка клинико-лабораторных показателей;
г) параметры жизненно важных функций организма участников клинического исследования, данные объективного исследования и другая информация обследования, которая касается вопросов безопасности.
12) в виде приложений к отчету представляются следующие сведения:
а) таблицы, рисунки, графики, на которые приводятся ссылки в отчете, но которые не вошли в текст отчета;
б) протокол клинического исследования и поправки к нему;
в) образец индивидуальной регистрационной карты;
г) перечень этических комитетов;
д) образцы письменной информации для пациентов и формы информированного согласия;
е) перечень и характеристики исследователей и других ответственных лиц;
ж) аналитическая документация, в случае использования в исследовании более одной серии исследуемого лекарственного препарата — перечни кодов пациентов, получавших лекарственный препарат разных серий;
з) схема рандомизации и коды (идентификация субъектов клинического исследования и назначенное лечение);
и) данные аудита (если проводились);
к) документация по статистическим методам;
л) документация по лабораторной стандартизации методов и обеспечения качества процедур, если применялись;
м) публикации, на которых базируется проведенное клиническое исследование.
II. Независимый этический комитет
10. Независимый этический комитет, созданный на уровне медицинской организации (локальный этический комитет), региональном уровне и функционирующий как независимый орган обеспечивает защиту прав, безопасность и охрану здоровья участников клинического исследования (далее — независимый этический комитет).
11. В состав независимого этического комитета должно входить достаточное число лиц, обладающих необходимым опытом и квалификацией для экспертной оценки научных, медицинских и этических аспектов планируемого клинического исследования, как правило, пять лиц, при этом интересы не менее чем одного лица должны лежать вне сферы науки.
12. Независимый этический комитет осуществляют свою деятельность в соответствии с утверждаемыми им стандартными операционными процедурами, содержащими требования, в том числе к составу и квалификации членов, сведения об учредителе, порядок организации проведения заседаний, рассмотрения документов и принятия по ним решений.
13. Независимый этический комитет рассматривает и принимает решение на основании следующих документов:
а) протокола клинического исследования;
б) брошюры исследователя;
в) информационного листка пациента;
г) сведений об опыте работы исследователей по соответствующим
специальностям и их опыте работы по проведению клинических исследований;
д) сведений о медицинских организациях, в которых предполагается проведение клинического исследования (полное и сокращенное наименования, организационно-правовая форма, место нахождения и место осуществления деятельности, телефон, телефакс, адрес электронной почты каждой медицинской организации);
е) сведений о предполагаемых сроках проведения клинического исследования;
ж) копии договора обязательного страхования, заключенного в соответствии с типовыми правилами обязательного страхования, с указанием предельной численности пациентов, участвующих в клиническом исследовании;
з) информации о составе лекарственного препарата.
Для рассмотрения независимым этическим комитетом заявителем могут быть предоставлены другие документы и материалы клинического исследования, в том числе материалы, содержащие описание действий, направленных на привлечение пациентов, здоровых добровольцев к участию в клиническом исследовании; письменные материалы, которые будут предоставлены участникам клинического исследования; информация о выплатах и компенсациях участникам клинического исследования; текущая версия научной биографии исследователя и (или) другие материалы, подтверждающие его квалификацию.
14. По результатам рассмотрения документов, указанных в пункте 13 настоящих Правил, независимый этический комитет принимает одно из следующих решений:
а) выдает заключение об одобрении проведения клинического исследования;
б) выдает заключение о невозможности одобрения клинического исследования;
в) рекомендует внести изменения в представленные документы для целей последующей выдачи заключения об одобрении проведения клинического исследования;
г) отменяет или приостанавливает ранее выданное заключение об одобрении проведения клинического исследования.
15. Независимый этический комитет:
а) осуществляет контроль за соблюдением этических норм при проведении клинического исследования и прав участников клинического исследования;
б) на основании научной биографии исследователя и иной документации оценивает соответствие квалификации исследователя планируемому клиническому исследованию;
в) в процессе исследования периодически рассматривает документацию и оценивает проводимое клиническое исследование (не реже одного раза в год);
г) может потребовать от организатора клинического исследования предоставить участнику клинического исследования дополнительные сведения об исследовании, помимо информации, содержащейся в информационном листке пациента, если по мнению независимого этического комитета, это позволит повысить степень защиты прав и безопасности участника клинического исследования;
д) оценивает размер и порядок осуществления выплат участникам клинического исследования с целью выявления необоснованной заинтересованности участников клинического исследования или принуждения их к участию в клиническом исследовании. Информация, касающаяся выплат участникам клинического исследования, включая методы, суммы и график выплат, отражается в информационном листке пациента.
е) может привлекать для принятия решений лиц, обладающих специальными знаниями в соответствующих областях, которые не участвуют в прениях и голосовании;
ж) согласовывает поправки в протокол клинического исследования;
з) совершает иные действия, направленные на исполнение своих функций и полномочий.
16. Независимый этический комитет незамедлительно в письменном виде сообщает исследователю, организатору клинического исследования о своих решениях, касающихся клинического исследования и причинах принятия решений.
17. Если протокол предусматривает невозможность получения согласия на участие в исследовании у пациента или его законного представителя до момента включения пациента в исследование, в том числе при неотложных состояниях независимый этический комитет должен убедиться, в том что представленный протокол и (или) другая документация соответствуют этическим нормам, а также иным обязательным требованиям для таких исследований.
18. Независимый этический комитет обеспечивает хранение документов, связанных с проведением клинического исследования, как правило, в течение трех лет после завершения клинического исследования и представление таких документов третьим лицам с соблюдением требований законодательства Российской Федерации о персональных данных, коммерческой, государственной и иной охраняемой законом тайне.
III. Организация, осуществляющая проведение клинического исследования
19. Организацию проведения клинических исследований для медицинского применения вправе осуществлять (далее — организатор клинического исследования):
а) разработчик лекарственного препарата или уполномоченное им лицо;
б) образовательные организации высшего образования, организации дополнительного профессионального образования;
в) научно-исследовательские организации.
20. Клинические исследования лекарственных препаратов проводятся по протоколу, разработанному организатором клинического исследования или привлеченным им юридическим лицом. Организатором клинического исследования могут вноситься изменения в протокол в форме поправок, оформленных в письменном виде посредством описания изменений или официальных разъяснений протокола.
21. Для получения разрешения на проведение клинического исследования лекарственного препарата организатор клинического исследования предоставляет в Министерство документы, предусмотренные в части 2 статьи 39 Федерального закона «Об обращении лекарственных средств».
22. Организатор клинического исследования:
а) до начала клинического исследования получает разрешение Министерства на проведение клинического исследования;
б) устанавливает и распределяет права, обязанности и ответственность всех лиц, участвующих в клиническом исследовании;
в) утверждает документы по порядку проведения клинического исследования, сбору, регистрации и представления данных в соответствии с протоколом и настоящими Правилами (далее — стандартные операционные процедуры организатора);
г) пересматривает брошюру не реже одного раза в год и, при необходимости, дополняет новыми данными, предоставляет актуальную редакцию брошюры исследователю (соисследователю), независимому этическому комитету;
д) при проведении клинического исследования осуществляет внедрение и поддержание систем обеспечения и контроля качества в соответствии со стандартными операционными процедурами организатора;
е) обеспечивает согласие всех привлеченных к участию в клиническом исследовании сторон на предоставление прямого доступа во все участвующие в клиническом исследовании медицинские организации ко всем первичным данным (или) документам и отчетам, полученным и составленным при проведении клинического исследования, в целях мониторинга и аудита качества проведения клинического исследования;
ж) назначает лиц, обладающих необходимыми знаниями и квалификацией, которые оказывают консультативную помощь исследователями по вопросам медицинского характера при проведении клинического исследования;
з) использует, присвоенный исследователем каждому участнику клинического исследования идентификационный код (уникальный номер, состоящий из цифр и (или) буквенных обозначений, используемый вместо фамилии, имени, отчества (при наличии) участника клинического исследования в отчетах о нежелательных явлениях, нежелательных реакциях и других данных для обеспечения конфиденциальности его личных данных и позволяющий идентифицировать все данные по каждому участнику);
и) обеспечивает ведение индивидуальной регистрационной карты в отношении каждого участника клинического исследования (на бумажном носителе или в форме электронного документа), куда вносится вся информация в соответствии с протоколом (далее — индивидуальная регистрационная карта);
к) принимает решение об образовании независимого комитета по мониторингу данных для оценки проводимого клинического исследования, рассмотрения данных по безопасности и эффективности исследуемого лекарственного препарата, в том числе с целью выработки рекомендаций о целесообразности продолжения, прекращения клинического исследования или внесения в изменений в протокол;
л) обеспечивает хранение документов, относящихся к клиническому исследованию, при прекращении клинической разработки исследуемого лекарственного препарата по одному, нескольким или всем показаниям, путям введения, лекарственным формам, как правило, в течение двух лет с момента официального прекращения разработки;
м) при прекращении клинической разработки исследуемого лекарственного препарата сообщает об этом всем участвующим в клиническом исследовании исследователям и медицинским организациям, в которых осуществляется проведение такого исследования;
н) сообщает о передаче прав на данные об исследуемом лекарственном препарате в Министерство;
о) осуществляет выбор исследователей и медицинских организаций для проведения клинического исследования, при проведении многоцентрового клинического исследования может быть организован координационный комитет и (или) выбран координатор из числа исследователей;
п) на всех этапах клинического исследования (от разработки протокола, индивидуальной регистрационной карты, плана статистического анализа, для общего руководства клиническим исследованием, работы с данными, верификации данных — до проведения статистического анализа полученных данных и подготовки промежуточного и финального отчетов о клиническом исследовании) привлекает лиц, обладающих соответствующей квалификацией;
р) обеспечивает контроль за качеством и полнотой полученных в ходе клинического исследования данных.
23. При использовании электронных систем для работы с данными клинического исследования и (или) электронных систем удаленного доступа к указанным данным организатор клинических исследований:
а) обеспечивает и документально оформляет соответствие систем электронной обработки данных требованиям к полноте, точности и надежности данных, а также стабильность достижения требуемого результата (далее — валидация данных);
б) утверждает стандартные операционные процедуры использования электронных систем;
в) обеспечивает работу электронных систем таким образом, чтобы при изменении введенных данных вносимые изменения были задокументированы и ранее введенные данные не были удалены;
г) обеспечивает систему защиты данных клинического исследования, предотвращающую несанкционированный доступ к данным, в том числе посредством утверждения списка лиц, имеющих доступ к данным клинического исследования с правом внесения в них изменений и резервного копирования данных;
д) обеспечивает сохранность маскировки клинического исследования, проводимого слепым методом, при вводе и обработке данных в электронной системе.
24. Организатор клинического исследования предоставляет исследователю и медицинской организации протокол клинического исследования и брошюру в текущей редакции до подписания с медицинской организацией договора на проведение клинического исследования и дает время для ознакомления с предоставленной информацией.
25. Организатор клинического исследования должен получить письменное согласие исследователя и уполномоченного лица медицинской организации, посредством подписания протокола или другого документа на:
а) проведение клинического исследования в соответствии с протоколом, настоящими Правилами и требованиями законодательства Российской Федерации в сфере обращения лекарственных средств;
б) соблюдение процедуры регистрации и представления данных клинического исследования;
в) проведение мониторинга и аудита;
г) хранение документов, связанных с проведением клинического исследования, до тех пор, пока организатор клинического исследования не сообщит исследователю и медицинской организации, что данные документы могут быть уничтожены.
26. Организатор клинического исследования обязан в качестве страхователя страховать риск причинения вреда жизни, здоровью пациента в результате проведения клинического исследования лекарственного препарата для медицинского применения за свой счет путем заключения договора обязательного страхования. Условия договора страхования, размер и порядок страховой выплаты устанавливаются в соответствии с постановлением № 714.
27. При наличии в медицинской организации, в которой планируется проведение клинического исследования, независимого этического комитета, организатор клинического исследования должен получить подтверждение того, что данный независимый этический комитет организован и действует в соответствии с настоящими Правилами и согласие этого независимого этического комитета на проведение клинического исследования.
В случае если согласие независимого этического комитета на проведение клинического исследования выражается посредством внесения изменений в документы и данные клинического исследования, медицинская организация предоставляет организатору клинического исследования копии всех измененных документов и данных.
В случае изменения решения независимого этического комитета относительно проведения клинического исследования, в том числе посредством отзыва ранее данного согласия, медицинская организация незамедлительно уведомляет об этом организатора клинического исследования.
28. Организатор клинического исследования должен иметь данные по безопасности и эффективности исследуемого лекарственного препарата, обосновывающие его применение, а также обновлять брошюру по мере получения в рамках проведения клинического исследования новых данных, влияющих на эффективность и безопасность исследуемого лекарственного препарата.
29. Организатор клинического исследования обеспечивает использование при проведении клинического исследования исследуемых лекарственных препаратов (включая, препараты сравнения и плацебо) произведенных в соответствии с требованиями надлежащей производственной практики, имеющих соответствующие показатели качества и которые в соответствующих случаях закодированы и маркированы в целях обеспечения маскировки.
На первичную упаковку (если для этого существует техническая возможность) и вторичную (потребительскую) упаковку лекарственных препаратов, предназначенных для клинических исследований, должна наноситься надпись: «Для клинических исследований».
30. Для клинических исследований, проводимых слепым методом, система кодирования исследуемого лекарственного препарата должна включать в себя механизм, позволяющий в экстренных случаях, быстро идентифицировать данный лекарственный препарат, при этом не допускающий возможности незаметно раскрыть код.
31. Если лекарственная форма исследуемого лекарственного препарата или препарата сравнения была изменена в ходе клинического исследования, то до использования новой лекарственной формы в клиническом исследовании должны быть получены результаты соответствующих дополнительных исследований данной лекарственной формы препарата, необходимые для оценки степени влияния данных изменений на фармакокинетику исследуемого лекарственного препарата, его безопасность и эффективность. В таком случае организатор клинического исследования обеспечивает внесение изменений в протокол.
32. Организатор клинического исследования обеспечивает поступление в медицинскую организацию, в которой проводится клиническое исследование, исследуемого лекарственного препарата.
Организатор клинического исследования должен иметь исследуемый лекарственный препарат в количестве, необходимом для проведения клинического исследования и подтверждения его соответствия требованиям фармакопейных статей либо в случае их отсутствия нормативной документации или нормативного документа, а также вести учет анализов и характеристик образцов лекарственного препарата из партии (серии).
Поступление исследуемого лекарственного препарата в медицинские организации, его возврат, уничтожение исследуемого лекарственного препарата подробно и в срок документируется организатором клинического исследования.
Организатор клинического исследования осуществляет постоянную оценку безопасности исследуемого лекарственного препарата и уведомляет всех занятых в клиническом исследовании исследователей и медицинские организации о полученных данных, которые могут неблагоприятно отразиться на безопасности участников клинического исследования и (или) повлиять на проведение клинического исследования.
33. Организатор клинического исследования сообщает всем участвующим в клиническом исследовании исследователям, медицинским организациям и Федеральной службе по надзору в сфере здравоохранения обо всех серьезных и непредвиденных нежелательных реакциях, а также представляет в Федеральную службу по надзору в сфере здравоохранения периодические отчеты по безопасности исследуемого препарата в установленном порядке.
34. Организатор клинического исследования осуществляет мониторинг клинического исследования, включающий деятельность по контролю за ходом клинического исследования, по обеспечению его проведения, сбору данных и представлению результатов в соответствии с протоколом, стандартными операционными процедурами, настоящими Правилами и требованиями законодательства Российской Федерации об обращении лекарственных средств.
Для проведения мониторинга организатор клинического исследования назначает физическое лицо, обладающее научными и (или) специальными знаниями, необходимыми для проведения мониторинга клинического исследования.
Лицо, назначенное организатором клинического исследования для проведения мониторинга, обязано соблюдать стандартные операционные процедуры организатора клинического исследования.
35. Целью мониторинга клинического исследования является установление того, что:
а) права и благополучие участников клинического исследования защищены;
б) полученные в ходе клинического исследования данные являются точными, полными и подтверждаются первичной документацией;
в) клиническое исследование проводится в соответствии с утвержденной текущей версией протокола и изменений к нему, требованиями настоящих Правил и законодательства Российской Федерации об обращении лекарственных средств.
36. Для достижения целей мониторинга клинического исследования лицо, назначенное организатором клинического исследования для проведения мониторинга:
а) обеспечивает взаимодействие между организатором клинического исследования, медицинской организацией и исследователем;
б) проверяет и контролирует наличие квалификации исследователя и ресурсов медицинской организации необходимых для проведения клинического исследования, включая лаборатории, оборудование и персонал;
в) в отношении исследуемого лекарственного препарата проверяет:
сроки и условия хранения, количество в объеме, необходимом для проведения клинического исследования;
получение только теми участниками клинического исследования, которым он назначен, и в дозах, установленных протоколом;
инструкции по правильному применению, хранению и порядку возврата предоставлены участникам клинического исследования;
осуществление исследователем контроля и документального оформления получения, применения, возврата, уничтожения исследуемого лекарственного препарата в медицинской организации;
г) проверяет соблюдение исследователем утвержденного протокола и всех изменений к нему, правильность, полноту, точность и сроки оформления исследователем документов и данных клинического исследования;
д) проверяет получение добровольного письменного согласия каждого участника клинического исследования до начала его участия в клиническом исследовании;
е) обеспечивает наличие у исследователя текущей редакции брошюры, иных документов и материалов, необходимых для проведения клинического исследования;
ж) обеспечивает доведение до исследователей необходимой для проведения клинического исследования информации;
з) проверяет соблюдение исследователем критериев отбора участников клинического исследования;
и) проверяет правильность, полноту и сроки регистрации данных клинического исследования, а также порядок ведения документов клинического исследования;
к) проверяет правильность и полноту регистрации в индивидуальной регистрационной карте данных, предусмотренных протоколом, данных об изменениях дозы и (или) терапии, о выявленных нежелательных явлениях и нежелательных реакциях, о пропущенных участником клинического исследования визитах, о невыполненных анализах и обследованиях. Все случаи исключения и выбывания участников клинического исследования из клинического исследования подлежат регистрации и пояснению в индивидуальной регистрационной карте;
л) сообщает исследователю о любых допущенных в индивидуальной регистрационной карте ошибках, пропусках и неразборчивых записях и прослеживает за тем, чтобы соответствующие исправления, были сделаны, датированы, объяснены (если необходимо) и подписаны исследователем;
м) сообщает исследователю об отклонениях от протокола, стандартных письменных процедур организатора, требований настоящих Правил, законодательства Российской Федерации об обращении лекарственных средств и принимает меры по их устранению и недопущению повторения.
37. Письменные отчеты по мониторингу клинического исследования представляются лицом, назначенным для проведения мониторинга организатору клинического исследования в порядке и сроки, установленные стандартными операционными процедурами организатора.
Отчет по мониторингу клинического исследования должен содержать, в том числе сроки проведения мониторинга, наименование медицинской организации, где проводился мониторинг, данные об исследователях, участниках клинического исследователя, с которыми общалось (взаимодействовало) лицо, назначенное для проведения мониторинга, описание проверки, сведения о выявленных недостатках, выводы по результатам мониторинга и иные сведения и данные, касающиеся мониторинга клинического исследования.
38. Организатором клинического исследования клинического исследования осуществляется независимая от мониторинга оценка соответствия проводимого клинического исследования протоколу, стандартным операционным процедурам организатора, настоящим Правилам и требованиям законодательства Российской Федерации об обращении лекарственных средств (далее — аудит клинического исследования).
Для проведения аудита клинического исследования организатор клинического исследования назначает лиц, независимых от всех субъектов клинического исследования и обладающих квалификацией, подготовкой, опытом для проведения аудита.
План и объем аудита клинического исследования разрабатываются и утверждаются организатором клинического исследования с учетом количества участников клинического исследования, типа и сложности клинического исследования, степени риска для участников клинического исследования и иных обстоятельств.
39. При обнаружении в ходе мониторинга или аудита клинического исследования серьезных и (или) повторяющихся случаев несоблюдения исследователем, медицинской организацией установленных требований к проведению клинического исследования организатор клинического исследования прекращает участие исследователя, медицинской организации в клиническом исследовании и вносит изменения в протокол.
40. В срок, не превышающий пяти рабочих дней со дня завершения, приостановления или прекращения клинического исследования лекарственного препарата для медицинского применения, сообщение об этом направляется организатором клинического исследования в Министерство.
41. При многоцентровых клинических исследованиях организатор клинического исследования обеспечивает:
а) проведение клинического исследования всеми медицинскими организациями, участвующими в многоцентровом клиническом исследовании, в строгом соответствии с протоколом клинического исследования;
б) разработку индивидуальных регистрационных карт, позволяющих собрать требуемые данные из всех медицинских организаций, участвующих в многоцентровом клиническом исследовании;
в) документальное закрепление прав и обязанностей медицинских организаций и исследователей, предоставление указанным лицам протокола, стандартных операционных процедур организатора, инструкции по заполнению индивидуальных регистрационных карт до начала клинического исследования.
IV. Организация работы исследователя
42. Руководитель медицинской организации, которая проводит клиническое исследование, назначает исследователя, ответственного за проведение такого исследования и имеющего лечебную специальность, соответствующую проводимому клиническому исследованию, со стажем работы по программам клинических исследований не менее чем три года и по его предложению назначает соисследователей из числа врачей этой медицинской организации (далее — исследователь).
43. Исследователь, соисследователи должны быть ознакомлены с результатами доклинического исследования исследуемого лекарственного препарата, актуальной версией брошюры, протоколом, иными документами и данными, имеющими отношение к проведению клинического исследования.
44. Исследователь осуществляет отбор участников клинического исследования, обеспечивает оказание медицинской помощи участнику клинического исследования.
45 Исследователь, соисследователи должны знать и соблюдать настоящие Правила и иные требования законодательства Российской Федерации об обращении лекарственных средств. Нарушение настоящих Правил, фальсификация результатов клинического исследования влекут за собой ответственность в соответствии с законодательством Российской Федерации.
46. Исследователь должен располагать временем и ресурсами, включая лаборатории, оборудование и персонал, необходимыми для проведения клинического исследования.
47. При наличии согласия участника клинического исследования исследователь сообщает лечащему врачу участника клинического исследования об участии последнего в клиническом исследовании.
48. Исследователь проводит клиническое исследование в соответствии с протоколом.
Исследователь обязан соблюдать протокол, не должен вносить в него изменения без решения Министерства и одобрения независимого этического комитета за исключением случаев, когда требуется устранить непосредственную угрозу жизни и (или) здоровья участника клинического исследования.
Любое отклонение от утвержденного протокола оформляется документально и в кратчайшие сроки направляется для рассмотрения и согласования в независимый этический комитет и организатору клинического исследования для согласования.
49. Исследователь сообщает в независимый этический комитет:
а) об отклонениях от протокола или изменениях протокола, произведенных для устранения угрозы жизни и (или) здоровья участника клинического исследования;
б) об изменениях, непосредственно влияющих на проведение клинического исследования и (или) увеличивающих риск участия в клиническом исследовании;
в) обо всех нежелательных реакциях, которые являются одновременно серьезными и непредвиденными;
г) о новых данных, которые могут свидетельствовать о возрастании риска для участников клинического исследования или могут неблагоприятно повлиять на ход клинического исследования.
50. Исследователь должен обеспечить применение участниками клинического исследования исследуемых лекарственных препаратов в соответствии с протоколом и соблюдать предусмотренную протоколом методику рандомизации (распределения участников клинического исследования по группам лечения или контроля случайным образом, позволяющего свести к минимуму субъективность) и обеспечить раскрытие кода только в соответствии с протоколом.
Если клиническое исследование проводится слепым методом исследователь документально оформляет и объясняет организатору клинического исследования любое преждевременное раскрытие кода исследуемых лекарственных препаратов.
51. Исследователь осуществляет учет исследуемых лекарственных препаратов и препаратов сравнения, в том числе ведет учет их поступлений, фактического наличия, количества использования каждым участником клинического исследования, уничтожения, а также возврата организатору клинического исследования.
Исследователь может передать обязанности по учету исследуемых лекарственных препаратов и препаратов сравнения работнику аптечной организации или иному лицу, подконтрольному исследователю.
Записи по учету должны включать в себя даты, количество, номера партий, серий, сроки годности (где применимо) и уникальные коды исследуемых лекарственных препаратов и препаратов сравнения и участников клинического исследования.
Исследователь должен вести записи, подтверждающие, что участники клинического исследования получали исследуемые лекарственные препараты и (или) препараты сравнения в дозах и количествах, предусмотренных протоколом клинического исследования.
52. Исследователь информирует участника клинического исследования или его законного представителя:
а) о том, что клиническое исследование носит экспериментальный характер, участие лица в клиническом исследовании является добровольным и оно может отказаться от участия в клиническом исследовании в любой момент;
б) о цели клинического исследования, его продолжительности и приблизительном количестве участников;
в) о вариантах лечения в процессе клинического исследования и вероятности случайного распределения в одну из групп лечения;
г) о процедурах клинического исследования, включая все инвазивные процедуры;
д) об обязанностях участника клинического исследования;
е) об ожидаемых риске и (или) пользе для участника клинического исследования, а также, в соответствующих случаях, для эмбриона, плода или грудного ребенка;
ж) об иных, помимо предусмотренных протоколом процедурах или методах лечения, которые могут быть доступны участнику клинического исследования, а также их потенциальных выгоде, пользе, риске;
з) о компенсации и (или) лечении, доступные участнику клинического исследования в случае причинения вреда его здоровью в результате участия в клиническом исследовании;
и) о планируемых выплатах участнику клинического исследования за его участие в клиническом исследовании, если таковые предусмотрены;
к) о планируемых расходах участника клинического исследования, если таковые ожидаются, связанные с его участием в клиническом исследовании;
л) о том, что участник клинического исследования или его законный представитель, подписывая информационный листок пациента, дает разрешение на доступ лицу, назначенному для проведения мониторинга, аудиторов, независимых этических комитетов, уполномоченных органов к медицинским записям участника клинического исследования;
м) о том, что записи, идентифицирующие участника клинического исследования, будут сохранены в тайне, раскрытие их допускается в соответствии с законодательством Российской Федерации и при публикации результатов клинического исследования конфиденциальность данных участника клинического исследования будет сохранена;
н) о том, что участник клинического исследования или его законный представитель будет незамедлительно ознакомлен с новой информацией, способной повлиять на его желание продолжать участие в клиническом исследовании;
о) о лицах, к которым можно обратиться для получения дополнительной информации о клиническом исследовании, и правах участников клинического исследования;
п) о возможных обстоятельствах и (или) причинах, по которым участие лица в клиническом исследовании может быть прекращено.
53. Информация о клиническом исследовании должна содержать как можно меньше специальных терминов и быть понятна участнику клинического исследования или его законному представителю.
54. Исследователь перед получением информированного добровольного согласия должен предоставить участнику клинического исследования, его законному представителю время, необходимое для принятия решения об участии в клиническом исследовании или отказе от такого участия. Участник клинического исследования или его законный представитель вправе получить исчерпывающие и достоверные ответы на все вопросы о клиническом исследовании.
55. Перед включением его в клиническое исследование участник клинического исследования, его законный представитель должен получить подписанный и датированный экземпляр информационного листка пациента и иные материалы, касающиеся проведения клинического исследования. В период проведения клинического исследования участнику клинического исследования сообщается обо всех изменениях в документы и данные клинического исследования, касающиеся его участия в клиническом исследовании.
56. В срок, установленный протоколом, исследователь обязан сообщать организатору клинического исследования обо всех серьезных нежелательных реакциях, за исключением тех, которые в протоколе или в брошюре определены как не требующие немедленного сообщения.
После первого сообщения о серьезных нежелательных реакциях исследователь в кратчайшие сроки представляет организатору клинического исследования подробный письменный отчет. Первый и последующие отчеты должны идентифицировать участников клинического исследования по присвоенным им уникальным кодам.
57. При сообщениях о смерти участника клинического исследования исследователь обязан по запросу организатора клинического исследования, независимого этического комитета, Министерства и (или) Федеральной службы по надзору в сфере здравоохранения предоставить любую дополнительную информацию относительно данного случая, в том числе протокол вскрытия и посмертный эпикриз.
58. В случае возникновения опасности для жизни, здоровья участника клинического исследования, исследователь обязан проинформировать об этом руководителя медицинской организации и организатора клинического исследования лекарственного препарата в течение 24 часов. Решение о приостановлении клинического исследования принимают руководитель медицинской организации и (или) организатор клинического исследования, решение о прекращении такого исследования принимает уполномоченный федеральный орган исполнительной власти на основании сообщения в письменной форме руководителя медицинской организации или организатор клинического исследования.
При досрочном прекращении клинического исследования или его приостановлении исследователь и (или) медицинская организация, в которой проводилось клиническое исследования обязаны незамедлительно информировать участников клинического исследования, обеспечить им необходимое лечение и наблюдение, проинформировать организатора клинического исследования, независимый этический комитет с предоставлением подробного письменного объяснения причин приостановления или прекращения клинического исследования.
59. Исследователь обеспечивает полное и достоверное ведение документов клинического исследования, включая записи на бумажных, электронных, магнитных или оптических носителях, сканограммы, рентгеновские снимки, электрокардиограммы, которые описывают методы, организацию и (или) результаты клинического исследования.
60. Хранение документов клинического исследования осуществляется медицинской организацией в соответствии с условиями договора, заключенного с организатором клинического исследования, или если данное условие не оговорено, как правило, в течение двух лет после государственной регистрации лекарственного препарата в Российской Федерации или официального прекращения клинической разработки исследуемого лекарственного препарата.
61. По завершении клинического исследования исследователь сообщает об этом руководителю медицинской организации, подготавливает отчет в соответствии с требованиями пункта 9 настоящих Правил и представляет его организатору клинического исследования и в независимый этический комитет.
Официальная публикация документа: Приказ Министерства здравоохранения Российской Федерации от 01.04.2016 № 200н «Об утверждении правил надлежащей клинической практики»
(Зарегистрирован 23.08.2016 № 43357)
Форма обратной связи
- О компании
- Качество
- Примеры переводов
- Карта сайта
- Вакансии
- Термины
- Блог
- Контакты
- Нормативные документы
- Конфиденциальность
- Обработка персональных данных
- Ограничение ответственности

![]()
Клинические испытания
лекарственного препарата и Правила надлежащей клинической практики GCP
Работу выполнили студентки 4 курса группы ХББО-02-16
Куменкова Анна и Иванова Елена

Клинические исследования
Клиническое исследование/испытание (КИ) –
это любой исследовательский проект, в котором проспективно разделяют участников на опытную и контрольную группы для изучения причинно-следственной связи между медицинским вмешательством и реакцией на него организма человека (определение Международного комитета
редакторов медицинских 2
журналов).

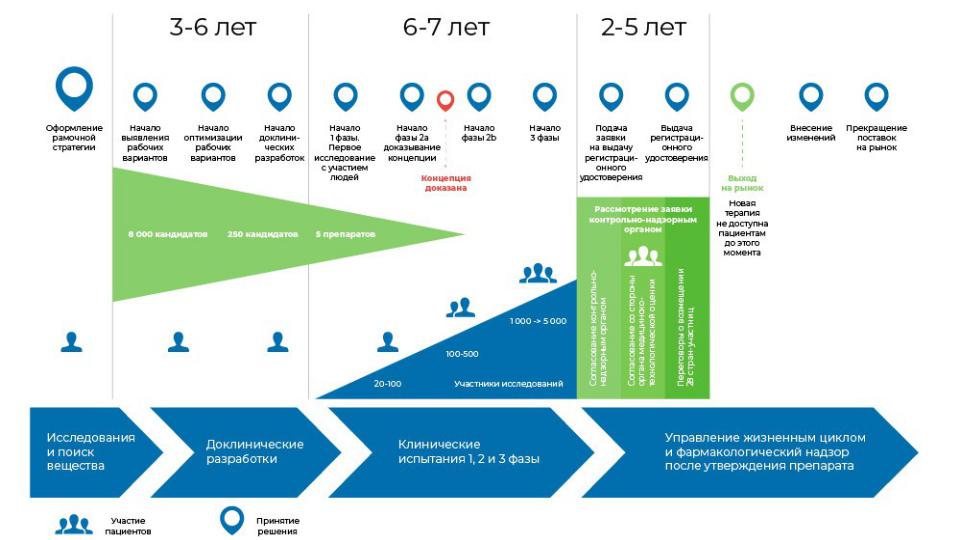
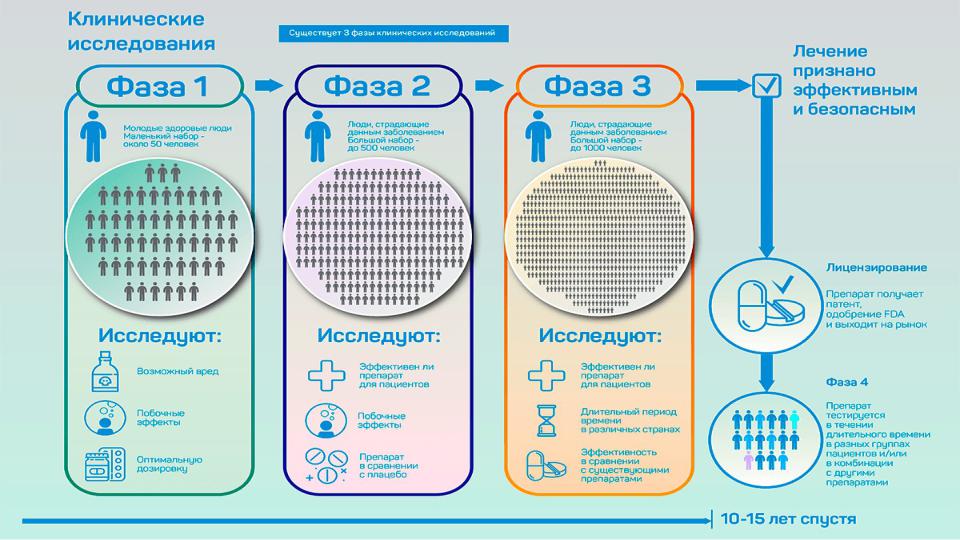
Зачем нужны КИ?
У людей Талидомид вызывает дефекты развития плода. В Европе, Австралии и Японии около 10 000 детей родились с
мальформациями
(пороками развития) конечностей. Препарат запретили в большинстве стран в 1961 году.
3
4
5

Что такое GCP?
Надлежащая клиническая практика (Good Clinical Practice; GCP) представляет собой международный этический и научный стандарт планирования и проведения исследований с участием человека в качестве субъекта, а также документального оформления и представления результатов таких исследований.
6

Для чего необходим стандарт?
Соблюдение указанного стандарта служит для общества гарантией того, что права, безопасность и благополучие субъектов исследования защищены, согласуются с принципами, заложенными Хельсинкской декларацией Всемирной медицинской ассоциации (ВМА), и что данные клинического исследования достоверны.
7

Принципы GCP
1) Клинические исследования должны проводиться в соответствии с этическими принципами, заложенными Хельсинкской декларацией ВМА и отраженными в настоящих правилахи нормативных требованиях.
2) До начала клинического исследования должна быть проведена оценка соотношения ожидаемой пользы к возможному риску для участника исследования и общества. Клиническое исследование может быть начато только в том случае, если ожидаемая польза оправдывает возможный риск.
3) Права, безопасность и благополучие участника клинического исследования имеют первостепенное значение и должны превалировать над интересами науки и общества.
4) Информация (доклиническая и клиническая) об исследуемом лекарственном препарате должна быть достаточной для обоснования предполагаемого
клинического исследования.
8

Принципы GCP
5)Клинические исследования должны отвечать научным требованиям и быть четко и подробно описаны в протоколе.
6)Клиническое исследование должно проводиться в соответствии с протоколом, утвержденным уполномоченным федеральным органом исполнительной власти, Советом по этике и Независимым этическим комитетом исследовательского центра при его наличии.
7)Ответственность за оказываемую участнику клинического исследования медицинскую помощь и принятие решений медицинского характера несет квалифицированный врач, являющийся исследователем или соисследователь, которому эта функция делегирована исследователем .
 Все привлекаемые к проведению клинического исследования лица должны иметь соответствующие образование, подготовку и опыт для выполнения
Все привлекаемые к проведению клинического исследования лица должны иметь соответствующие образование, подготовку и опыт для выполнения
|
возложенных на них задач. |
9 |
9) Добровольное информированное согласие должно быть получено у каждого

Принципы GCP
10)Всю полученную в клиническом исследовании информацию необходимо регистрировать, передавать и хранить таким образом, чтобы были обеспечены точность и правильность ее представления, интерпретации и верификации.
11)Конфиденциальность записей, позволяющих идентифицировать субъектов исследования, должна быть обеспечена с соблюдением права на частную жизнь и защиту конфиденциальности в соответствии с нормативными требованиями.
12)Производство и хранение исследуемых препаратов, а также обращение с ними необходимо осуществлять в соответствии с правилами надлежащей производственной практики. Исследуемые препараты должны применяться в соответствии с утвержденным протоколом.
13)Для обеспечения качества каждого аспекта исследования должны быть внедрены соответствующие системы и операционные процедуры.
10
Зарегистрировано в Минюсте России 23 августа 2016 г. N 43357
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРИКАЗ
от 1 апреля 2016 г. N 200н
ОБ УТВЕРЖДЕНИИ ПРАВИЛ НАДЛЕЖАЩЕЙ КЛИНИЧЕСКОЙ ПРАКТИКИ
В соответствии с пунктом 18 статьи 5 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств» (Собрание законодательства Российской Федерации, 2010, N 16, ст. 1815; 2012, N 26, ст. 3446; 2013, N 27, ст. 3477; 2014, N 52 ст. 7540) и пунктом 5.2.155 Положения о Министерстве здравоохранения Российской Федерации, утвержденного постановлением Правительства Российской Федерации от 19 июня 2012 N 608 (Собрание законодательства Российской Федерации, 2012, N 26, ст. 3526; 2015, N 23, ст. 3333), приказываю:
1. Утвердить прилагаемые Правила надлежащей клинической практики.
2. Установить, что Правила надлежащей клинической практики, утвержденные настоящим приказом, распространяются на правоотношения по проведению клинических исследований лекарственного препарата для медицинского применения, заявления о выдаче разрешений на проведение которых поданы в установленном порядке после вступления в силу настоящего приказа.
3. Признать утратившим силу приказ Министерства здравоохранения Российской Федерации от 19 июня 2003 г. N 266 «Об утверждении Правил клинической практики в Российской Федерации» (зарегистрирован Министерством юстиции Российской Федерации 20 июня 2003 г., регистрационный N 4808).
Врио Министра
И.Н. КАГРАМАНЯН
УТВЕРЖДЕНЫ
приказом Министерства здравоохранения
Российской Федерации
от 1 апреля 2016 г. N 200н
ПРАВИЛА НАДЛЕЖАЩЕЙ КЛИНИЧЕСКОЙ ПРАКТИКИ
I. Общие положения
1. Настоящие Правила надлежащей клинической практики (далее — Правила) регулируют отношения по планированию, организации, проведению, мониторингу, аудиту, документированию клинических исследований лекарственных препаратов для медицинского применения (далее соответственно — клиническое исследование, лекарственный препарат) с участием человека в качестве субъекта клинического исследования, анализу и представлению результатов клинических исследований, обеспечивающие гарантию достоверности и точности полученных данных и представленных результатов, а также защиту прав, здоровья и конфиденциальности персональных данных участников клинического исследования.
2. Для целей настоящих Правил применяются термины и определения, используемые в Федеральном законе от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств» <1> (далее — Федеральный закон «Об обращении лекарственных средств»).
<1> Собрание законодательства Российской Федерации, 2010, N 16, ст. 1815; N 31, ст. 4161; N 42, ст. 5293; N 49, ст. 6409; 2011, N 50, ст. 7351; 2012, N 26, ст. 3446; N 53, ст. 7587; 2013, N 27, ст. 3477; N 48, ст. 6165; 2014, N 11, ст. 1098; N 43, ст. 5797; N 52, ст. 7540; 2015, N 10, ст. 1404; N 27, ст. 3951; N 29, ст. 4359, ст. 4367, ст. 4388; N 51, ст. 7245; 2016, N 2, ст. 325; N 9, ст. 1268; N 23, ст. 3287.
3. Объектом клинического исследования является лекарственная форма фармакологически активного вещества или плацебо, изучаемые или используемые для контроля в клиническом исследовании, или зарегистрированный лекарственный препарат для медицинского применения в случае, если способ его применения отличается от утвержденного (в рамках процедуры государственной регистрации), а также при его использовании по новому показанию или для получения дополнительной информации по утвержденному показанию (далее — исследуемый лекарственный препарат). Исследуемые лекарственные препараты применяются в соответствии с утвержденным (в рамках процедуры получения разрешения на проведение клинического исследования) протоколом клинического исследования.
Обращение исследуемых лекарственных препаратов осуществляется в соответствии с правилами надлежащей производственной практики и правилами надлежащей практики хранения и перевозки лекарственных препаратов <1>.
<1> Пункт 18 статьи 5 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
4. Клиническое исследование проводится в отношении физического лица (пациента или здорового добровольца), принимающего участие в клиническом исследовании в составе группы, получающей исследуемый лекарственный препарат, либо в составе контрольной группы (далее — участник клинического исследования).
Каждый участник клинического исследования после получения информации о клиническом исследовании и до начала проведения клинического исследования дает добровольное согласие на участие в таком клиническом исследовании посредством подписания информационного листка пациента.
Согласие на участие в клиническом исследовании может быть дано законным представителем <1> участника клинического исследования.
<1> В отношении лица, указанного в части 2 статьи 20 Федерального закона от 21 ноября 2011 г. N 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011, N 48, ст. 6724; 2013, N 48, ст. 6165).
5. Клиническое исследование лекарственного препарата проводится на основании разрешения на проведение клинического исследования, выданного Министерством здравоохранения Российской Федерации (далее — Министерство) по результатам экспертизы документов для получения разрешения на проведение клинического исследования и этической экспертизы, предусмотренных статьей 39 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
6. Экспертиза документов, указанных в пункте 5 настоящих Правил, для получения разрешения на проведение клинического исследования лекарственного препарата проводится федеральным государственным бюджетным учреждением по проведению экспертизы лекарственных средств <1> , этическая экспертиза осуществляется Советом по этике <2> ,<3>.
<1> Приказ Министерства здравоохранения и социального Российской Федерации от 26 августа 2010 г. N 750н «Об утверждении правил проведения экспертизы лекарственных средств для медицинского применения и формы заключения комиссии экспертов» (зарегистрирован Министерством юстиции Российской Федерации 31 августа 2010 г., регистрационный N 18315) с изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 13 декабря 2012 г. N 1041н (зарегистрирован Министерством юстиции Российской Федерации 10 апреля 2013 г., регистрационный N 28082), от 3 апреля 2014 г. N 152н (зарегистрирован Министерством юстиции Российской Федерации 10 июня 2014 г., регистрационный N 32648).
<2> Приказ Министерства здравоохранения и социального Российской Федерации от 26 августа 2010 г. N 753н «Об утверждении порядка организации и проведения этической экспертизы возможности проведения клинического исследования лекарственного препарата для медицинского применения и формы заключения совета по этике» (зарегистрирован Министерством юстиции Российской Федерации 31 августа 2010 г., регистрационный N 18303).
<3> Приказ Минздрава России от 29 ноября 2012 г. N 986н «Об утверждении Положения о Совете по этике» (зарегистрирован Министерством юстиции Российской Федерации 7 февраля 2013 г., регистрационный N 26897).
7. Клиническое исследование проводится в соответствии с протоколом клинического исследования (далее — протокол), который содержит, в том числе:
1) название протокола, идентификационный номер протокола и дату утверждения организатором исследования. Новые версии протокола имеют номер версии и дату, поправки к протоколу — номер поправки и дату;
2) наименование и адрес организации, осуществляющей организацию проведения клинического исследования и осуществляющей мониторинг (если они различные);
3) фамилию, имя, отчество (при наличии) и должность лица, уполномоченного от имени организации, осуществляющей организацию проведения клинического исследования, подписывать протокол и поправки к протоколу;
4) фамилию, имя, отчество (при наличии), должность, адрес и номер телефона медицинского эксперта данного клинического исследования, назначенного организацией, осуществляющей организацию проведения клинического исследования;
5) фамилию, имя, отчество (при наличии) и должность исследователя, отвечающего за проведение клинического исследования, а также адреса и номера телефонов клинических центров;
6) фамилию, имя, отчество (при наличии), должность, адрес и номер телефона врача, отвечающего за принятие решений медицинского характера (если данное лицо не является исследователем);
7) наименования и адреса клинических лабораторий и других медицинских и (или) технических служб и организаций, принимающих участие в клиническом исследовании;
обоснование клинического исследования, включающее:
а) название и описание исследуемых лекарственных препаратов (включая плацебо и активный контроль);
б) сводное изложение результатов доклинических исследований и клинических исследований (если ранее проводились) исследуемых лекарственных препаратов;
в) краткое описание известных и потенциальных рисков и пользы применения исследуемого лекарственного препарата для участников клинического исследования;
г) описание и обоснование способа введения, дозировки, режима дозирования и курса лечения;
д) указание на то, что клиническое исследование будет проводиться в соответствии с протоколом клинического исследования и настоящими Правилами;
е) описание исследуемой популяции;
ж) ссылки на литературные источники и данные, имеющие значение для клинического исследования и представляющие собой обоснование клинического исследования;
9) цели и задачи клинического исследования;
10) описание общего плана (дизайна) клинического исследования, включающего:
а) указание основных и дополнительных (при наличии) исследуемых параметров, которые будут оцениваться в ходе клинического исследования;
б) описание типа дизайна проводимого клинического исследования (двойное слепое, плацебо-контролируемое, параллельное и иные виды исследований) и графическую схему дизайна клинического исследования, процедур и этапов клинического исследования;
в) описание мер, направленных на минимизацию, исключение субъективности, в том числе рандомизации, слепого метода (маскировки) — метода, при применении которого одной или нескольким участвующим в клиническом исследовании сторонам неизвестно, какое лечение назначено участнику клинического исследования (простой слепой метод предусматривает неосведомленность участников клинического исследования о назначенном им виде лечения, двойной слепой метод подразумевает неосведомленность о назначенном им виде лечения участников клинического исследования, исследователей, и, в некоторых случаях, лиц, выполняющих статистическую обработку данных);
г) описание используемого в клиническом исследовании лечения, дозировок и схемы применения исследуемых лекарственных препаратов, описание лекарственной формы, упаковки и маркировки исследуемых лекарственных препаратов;
д) продолжительность участия пациентов или здоровых добровольцев в клиническом исследовании, описание последовательности и продолжительности всех периодов клинического исследования, в том числе периода последующего наблюдения, если таковой предусмотрен;
е) описание правил остановки частей клинического исследования и (или) клинического исследования в целом, критериев исключения для отдельных участников клинического исследования;
ж) описание процедуры учета исследуемых лекарственных препаратов, плацебо и препаратов сравнения;
з) описание способов хранения рандомизационных кодов и процедуры их раскрытия;
и) перечень данных, регистрируемых в индивидуальной регистрационной карте (без предварительной записи в письменном или электронном виде) и рассматриваемых в качестве первичных данных;
11) критерии включения участников в клиническое исследование;
12) критерии невключения участников в клиническое исследование;
13) критерии исключения участников из клинического исследования (основания прекращения применения исследуемого лекарственного препарата, исследуемого лечения);
14) сведения о всех используемых в клиническом исследовании лекарственных препаратах, включая их названия, дозировки, частоту приема, пути и способы введения, продолжительность лечения, периоды последующего наблюдения для каждой группы участников клинического исследования, сведения о разрешенных для применения участниками клинического исследования лекарственных препаратах (включая неотложную терапию) и запрещенных для применения до и (или) во время клинического исследования лекарственных препаратах, способах контроля за соблюдением участниками клинического исследования условий приема лекарственных препаратов;
15) перечень параметров эффективности исследуемого лекарственного препарата и методы, сроки проведения оценки, регистрации и анализа параметров эффективности лекарственного препарата;
16) перечень параметров безопасности исследуемого лекарственного препарата и методы и сроки проведения оценки, регистрации и анализа параметров безопасности исследуемого лекарственного препарата;
17) требования к отчетам, процедуре регистрации и сообщениям о нежелательных явлениях и интеркуррентных заболеваниях;
18) метод и продолжительность наблюдения за участниками клинического исследования после возникновения нежелательных явлений;
19) описание статистических методов клинического исследования, включающих, в том числе:
а) сроки проведения промежуточного анализа;
б) планируемое количество участников клинического исследования с обоснованием размера выборки;
в) применяемый уровень значимости клинического исследования;
г) критерии прекращения клинического исследования;
д) процедуры учета отсутствующих, не подлежащих анализу и сомнительных данных;
е) процедуры сообщения о любых отклонениях от первоначального статистического плана (все отклонения от первоначального статистического плана описываются и обосновываются в протоколе и (или) финальном отчете о клиническом исследовании);
ж) процедуры отбора участников клинического исследования для анализа (все рандомизированные участники клинического исследования, все участники клинического исследования, получившие хотя бы одну дозу исследуемого лекарственного препарата, все участники, соответствующие критериям отбора, участники клинического исследования, данные которых пригодны для оценки, иные категории участников);
20) описание действий по контролю качества и обеспечению качества;
21) описание этических аспектов клинического исследования;
22) описание работы с данными и ведению записей;
23) описание порядка финансирования клинического исследования и страхования участников клинического исследования;
24) указание на возможность публикации результатов клинического исследования.
8. Сводное изложение результатов доклинических и клинических исследований приводится в брошюре исследователя (далее — брошюра), содержащей, в том числе следующие сведения:
1) наименование организации-разработчика лекарственного препарата, номер клинического исследования, химическое, международное непатентованное, группировочное и торговое наименования лекарственного препарата, дату составления брошюры, номер версии брошюры, номер и дату предыдущей редакции брошюры, а также уведомление о конфиденциальности содержащейся в ней информации, указываемые на титульном листе брошюры;
2) краткое резюме о физических, химических, фармацевтических, фармакологических, токсикологических, фармакокинетических, метаболических и клинических свойствах исследуемого лекарственного препарата, соответствующих стадии клинической разработки исследуемого лекарственного препарата, обоснование для изучения исследуемого лекарственного препарата, его ожидаемые профилактические, терапевтические или диагностические показания, общий подход к оценке исследуемого лекарственного препарата;
з) описание физических, химических и фармацевтических свойств и лекарственной формы исследуемого лекарственного препарата, его компонентов (включая химические и (или) структурные формулы), обоснование (при необходимости) состава лекарственной формы, включая вспомогательные вещества;
4) результаты доклинических исследований фармакологических свойств, токсичности, фармакокинетики и метаболизма исследуемого лекарственного препарата, с описанием, в том числе:
а) использованных видов животных, их количество и пол в каждой группе;
б) единиц измерения дозы, кратности и пути введения, длительности курса введения;
в) системного распределения, продолжительности последующего наблюдения после окончания введения лекарственного препарата;
г) характера и частоты фармакологических или токсических эффектов, их выраженности или степени тяжести, дозозависимости эффектов, времени до наступления, обратимость, продолжительность эффектов;
д) доклинической фармакологии;
е) фармакокинетики, метаболизма и распределения исследуемого лекарственного средства в тканях животных всех видов, на которых проводились исследования, включая сведения о всасывании, местной и системной биодоступности исследуемого лекарственного средства и его метаболитов, а также их связь с данными фармакологических и токсикологических исследований на животных;
ж) токсикологии (описание токсических эффектов исследуемого лекарственного средства, выявленных в исследованиях на животных разных видов (если применимо), включая сведения о токсичности при однократном и многократном введении, канцерогенности, специальных исследованиях (местнораздражающее и аллергизирующее действие), репродуктивной токсичности, генотоксичности (мутагенности).
5) описание эффектов применения исследуемого лекарственного препарата у человека, в том числе:
а) фармакокинетики (включая метаболизм, всасывание, связывание с белками плазмы, распределение и выведение) и биодоступности исследуемого лекарственного препарата, взаимодействия исследуемого лекарственного препарата (лекарственные взаимодействия, влияние приема пищи и иные взаимодействия), других имеющихся данных по фармакокинетике (результаты проведенных в рамках клинических испытаний фармакокинетических исследований на различных группах);
6) данных по безопасности, фармакодинамике, эффективности и дозозависимости эффектов исследуемого лекарственного препарата (и его метаболитов, если есть данные), полученных в ходе проведенных клинических исследований (с участием здоровых добровольцев и (или) пациентов) и интерпретация этих данных (по законченным клиническим исследованиям представляются сводные отчеты по эффективности и безопасности исследуемого лекарственного препарата, а также сводные таблицы нежелательных реакций по всем клиническим исследованиям, описываются значимые различия в характере и частоте нежелательных реакций);
в) пострегистрационного опыта применения исследуемого лекарственного препарата с указанием страны, в которых исследуемый лекарственный препарат был зарегистрирован и имеется в продаже;
6) инструкции для исследователя по диагностике и лечению возможных передозировок и нежелательных реакций при применении исследуемого лекарственного препарата, основанные на предыдущем клиническом опыте и фармакологических свойствах исследуемого лекарственного препарата, а также обобщенная информация по различным свойствам исследуемого лекарственного препарата.
9. Отчет о результатах клинического исследования (далее — отчет) составляется в письменной форме организацией, осуществляющей организацию проведения клинического исследования лекарственного препарата, на основании заключений медицинских организаций, проводивших это исследование и включает, в том числе:
1) титульную страницу, на которой указываются:
а) название отчета;
б) название лекарственного препарата;
в) изучаемое показание к применению лекарственного препарата;
г) краткое описание дизайна клинического исследования, наличие препарата сравнения, продолжительность клинического исследования, дозирование лекарственного препарата и контингент участников исследования;
д) наименование организации, осуществляющей организацию проведения клинического исследования;
е) идентификационный номер протокола клинического исследования;
ж) фазы клинического исследования;
з) даты начала и окончания клинического исследования;
и) дата составления отчета;
к) сведения об исследователе, руководителе медицинской организации, ответственном представителе организации, осуществляющей организацию проведения клинического исследования;
2) резюме — краткое описание проведенного клинического исследования с числовыми данными для иллюстрации результатов;
3) оглавление, включая перечень и расположение приложений, таблиц;
4) перечень сокращений и определение терминов, используемых в отчете;
5) названия медицинских организаций, в которых проводилось клиническое исследование, место нахождения, контактный телефон;
6) цели и задачи клинического исследования;
7) план проведенного клинического исследования, включающий, в том числе:
а) общий план (дизайн), план-описание клинического исследования и схематическое изображение этапов и процедур клинического исследования;
б) обоснование плана (дизайна) клинического исследования;
в) критерии выбора популяции;
г) назначенное лечение, идентификация исследуемых лекарственных препаратов, методы распределения участников клинического исследования по группам (рандомизация), дозы и время приема исследуемого лекарственных препаратов, предшествующая и сопутствующая терапия;
д) данные об эффективности и безопасности исследуемого лекарственного препарата (оценка и график определения показателей эффективности и безопасности);
е) способы обеспечения качества и достоверности полученных данных, включая данные аудита, результаты инспекций, если проводились;
ж) изменения, внесенные в протокол клиническое исследование;
сведения об участниках клинического исследования и распределении их по группам;
9) сведения об отклонениях от протокола клинического исследования;
10) оценку эффективности лекарственного препарата, в том числе:
а) совокупность данных, подлежавших анализу при проведении клинического исследования;
б) демографические и (или) другие исходные данные;
в) сведения о соблюдении участниками клинического исследования схемы лечения;
г) результаты оценки эффективности: статистические, аналитические данные, выводы относительно эффективности;
11) сведения о безопасности лекарственного препарата, в том числе:
а) нежелательные реакции (краткое резюме о нежелательных реакциях, их анализ, списки нежелательных реакций, которые наблюдались у всех участников клинического исследования);
б) смерть и другие серьезные нежелательные явления;
в) оценка клинико-лабораторных показателей;
г) параметры жизненно важных функций организма участников клинического исследования, данные объективного исследования и другая информация обследования, которая касается вопросов безопасности.
12) в виде приложений к отчету представляются следующие сведения:
а) таблицы, рисунки, графики, на которые приводятся ссылки в отчете, но которые не вошли в текст отчета;
б) протокол клинического исследования и поправки к нему;
в) образец индивидуальной регистрационной карты;
г) перечень этических комитетов;
д) образцы письменной информации для пациентов и формы информированного согласия;
е) перечень и характеристики исследователей и других ответственных лиц;
ж) аналитическая документация, в случае использования в исследовании более одной серии исследуемого лекарственного препарата — перечни кодов пациентов, получавших лекарственный препарат разных серий;
з) схема рандомизации и коды (идентификация субъектов клинического исследования и назначенное лечение);
и) данные аудита (если проводились);
к) документация по статистическим методам;
л) документация по лабораторной стандартизации методов и обеспечения качества процедур, если применялись;
м) публикации, на которых базируется проведенное клиническое исследование.
II. Независимый этический комитет
10. Независимый этический комитет, созданный на уровне медицинской организации (локальный этический комитет), региональном уровне и функционирующий как независимый орган обеспечивает защиту прав, безопасность и охрану здоровья участников клинического исследования (далее — независимый этический комитет).
11. В состав независимого этического комитета должно входить достаточное число лиц, обладающих необходимым опытом и квалификацией для экспертной оценки научных, медицинских и этических аспектов планируемого клинического исследования, как правило, пять лиц, при этом интересы не менее чем одного лица должны лежать вне сферы науки.
12. Независимый этический комитет осуществляют свою деятельность в соответствии с утверждаемыми им стандартными операционными процедурами, содержащими требования, в том числе к составу и квалификации членов, сведения об учредителе, порядок организации проведения заседаний, рассмотрения документов и принятия по ним решений.
13. Независимый этический комитет рассматривает и принимает решение на основании следующих документов:
а) протокола клинического исследования;
б) брошюры исследователя;
в) информационного листка пациента;
г) сведений об опыте работы исследователей по соответствующим специальностям и их опыте работы по проведению клинических исследований;
д) сведений о медицинских организациях, в которых предполагается проведение клинического исследования (полное и сокращенное наименования, организационно-правовая форма, место нахождения и место осуществления деятельности, телефон, телефакс, адрес электронной почты каждой медицинской организации);
е) сведений о предполагаемых сроках проведения клинического исследования;
ж) копии договора обязательного страхования, заключенного в соответствии с типовыми правилами обязательного страхования, с указанием предельной численности пациентов, участвующих в клиническом исследовании <1>;
<1> Постановление Правительства Российской Федерации от 13 сентября 2010 г. N 714 «Об утверждении Типовых правил обязательного страхования жизни и здоровья пациента, участвующего в клинических исследованиях лекарственного препарата» (Собрание законодательства Российской Федерации, 2010, N 38, ст. 4832; 2011, N 22, ст. 3171; 2012, N 37, ст. 5002; 2014, N 43, ст. 5892) (далее — постановление N 714).
з) информации о составе лекарственного препарата.
Для рассмотрения независимым этическим комитетом заявителем могут быть предоставлены другие документы и материалы клинического исследования, в том числе материалы, содержащие описание действий, направленных на привлечение пациентов, здоровых добровольцев к участию в клиническом исследовании; письменные материалы, которые будут предоставлены участникам клинического исследования; информация о выплатах и компенсациях участникам клинического исследования; текущая версия научной биографии исследователя и (или) другие материалы, подтверждающие его квалификацию.
14. По результатам рассмотрения документов, указанных в пункте 13 настоящих Правил, независимый этический комитет принимает одно из следующих решений:
а) выдает заключение об одобрении проведения клинического исследования;
б) выдает заключение о невозможности одобрения клинического исследования;
в) рекомендует внести изменения в представленные документы для целей последующей выдачи заключения об одобрении проведения клинического исследования;
г) отменяет или приостанавливает ранее выданное заключение об одобрении проведения клинического исследования.
15. Независимый этический комитет:
а) осуществляет контроль за соблюдением этических норм при проведении клинического исследования и прав участников клинического исследования;
б) на основании научной биографии исследователя и иной документации оценивает соответствие квалификации исследователя планируемому клиническому исследованию;
в) в процессе исследования периодически рассматривает документацию и оценивает проводимое клиническое исследование (не реже одного раза в год);
г) может потребовать от организатора клинического исследования предоставить участнику клинического исследования дополнительные сведения об исследовании, помимо информации, содержащейся в информационном листке пациента, если по мнению независимого этического комитета, это позволит повысить степень защиты прав и безопасности участника клинического исследования;
д) оценивает размер и порядок осуществления выплат участникам клинического исследования с целью выявления необоснованной заинтересованности участников клинического исследования или принуждения их к участию в клиническом исследовании. Информация, касающаяся выплат участникам клинического исследования, включая методы, суммы и график выплат, отражается в информационном листке пациента.
е) может привлекать для принятия решений лиц, обладающих специальными знаниями в соответствующих областях, которые не участвуют в прениях и голосовании;
ж) согласовывает поправки в протокол клинического исследования;
з) совершает иные действия, направленные на исполнение своих функций и полномочий.
16. Независимый этический комитет незамедлительно в письменном виде сообщает исследователю, организатору клинического исследования о своих решениях, касающихся клинического исследования и причинах принятия решений.
17. Если протокол предусматривает невозможность получения согласия на участие в исследовании у пациента или его законного представителя до момента включения пациента в исследование, в том числе при неотложных состояниях независимый этический комитет должен убедиться, в том что представленный протокол и (или) другая документация соответствуют этическим нормам, а также иным обязательным требованиям для таких исследований.
18. Независимый этический комитет обеспечивает хранение документов, связанных с проведением клинического исследования, как правило, в течение трех лет после завершения клинического исследования и представление таких документов третьим лицам с соблюдением требований законодательства Российской Федерации о персональных данных, коммерческой, государственной и иной охраняемой законом тайне.
III. Организация, осуществляющая проведение клинического исследования
19. Организацию проведения клинических исследований для медицинского применения вправе осуществлять (далее — организатор клинического исследования):
а) разработчик лекарственного препарата или уполномоченное им лицо;
б) образовательные организации высшего образования, организации дополнительного профессионального образования;
в) научно-исследовательские организации <1>.
<1> Часть 3 статьи 38 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
20. Клинические исследования лекарственных препаратов проводятся по протоколу, разработанному организатором клинического исследования или привлеченным им юридическим лицом. Организатором клинического исследования могут вноситься изменения в протокол в форме поправок, оформленных в письменном виде посредством описания изменений или официальных разъяснений протокола.
21. Для получения разрешения на проведение клинического исследования лекарственного препарата организатор клинического исследования предоставляет в Министерство документы, предусмотренные в части 2 статьи 39 Федерального закона «Об обращении лекарственных средств».
22. Организатор клинического исследования:
а) до начала клинического исследования получает разрешение Министерства на проведение клинического исследования;
б) устанавливает и распределяет права, обязанности и ответственность всех лиц, участвующих в клиническом исследовании;
в) утверждает документы по порядку проведения клинического исследования, сбору, регистрации и представления данных в соответствии с протоколом и настоящими Правилами (далее — стандартные операционные процедуры организатора);
г) пересматривает брошюру не реже одного раза в год и, при необходимости, дополняет новыми данными, предоставляет актуальную редакцию брошюры исследователю (соисследователю), независимому этическому комитету;
д) при проведении клинического исследования осуществляет внедрение и поддержание систем обеспечения и контроля качества в соответствии со стандартными операционными процедурами организатора;
е) обеспечивает согласие всех привлеченных к участию в клиническом исследовании сторон на предоставление прямого доступа во все участвующие в клиническом исследовании медицинские организации ко всем первичным данным (или) документам и отчетам, полученным и составленным при проведении клинического исследования, в целях мониторинга и аудита качества проведения клинического исследования;
ж) назначает лиц, обладающих необходимыми знаниями и квалификацией, которые оказывают консультативную помощь исследователями по вопросам медицинского характера при проведении клинического исследования;
з) использует, присвоенный исследователем каждому участнику клинического исследования идентификационный код (уникальный номер, состоящий из цифр и (или) буквенных обозначений, используемый вместо фамилии, имени, отчества (при наличии) участника клинического исследования в отчетах о нежелательных явлениях, нежелательных реакциях и других данных для обеспечения конфиденциальности его личных данных и позволяющий идентифицировать все данные по каждому участнику);
и) обеспечивает ведение индивидуальной регистрационной карты в отношении каждого участника клинического исследования (на бумажном носителе или в форме электронного документа), куда вносится вся информация в соответствии с протоколом (далее — индивидуальная регистрационная карта);
к) принимает решение об образовании независимого комитета по мониторингу данных для оценки проводимого клинического исследования, рассмотрения данных по безопасности и эффективности исследуемого лекарственного препарата, в том числе с целью выработки рекомендаций о целесообразности продолжения, прекращения клинического исследования или внесения в изменений в протокол;
л) обеспечивает хранение документов, относящихся к клиническому исследованию, при прекращении клинической разработки исследуемого лекарственного препарата по одному, нескольким или всем показаниям, путям введения, лекарственным формам, как правило, в течение двух лет с момента официального прекращения разработки;
м) при прекращении клинической разработки исследуемого лекарственного препарата сообщает об этом всем участвующим в клиническом исследовании исследователям и медицинским организациям, в которых осуществляется проведение такого исследования;
н) сообщает о передаче прав на данные об исследуемом лекарственном препарате в Министерство;
о) осуществляет выбор исследователей и медицинских организаций для проведения клинического исследования, при проведении многоцентрового клинического исследования может быть организован координационный комитет и (или) выбран координатор из числа исследователей;
п) на всех этапах клинического исследования (от разработки протокола, индивидуальной регистрационной карты, плана статистического анализа, для общего руководства клиническим исследованием, работы с данными, верификации данных — до проведения статистического анализа полученных данных и подготовки промежуточного и финального отчетов о клиническом исследовании) привлекает лиц, обладающих соответствующей квалификацией;
р) обеспечивает контроль за качеством и полнотой полученных в ходе клинического исследования данных.
23. При использовании электронных систем для работы с данными клинического исследования и (или) электронных систем удаленного доступа к указанным данным организатор клинических исследований:
а) обеспечивает и документально оформляет соответствие систем электронной обработки данных требованиям к полноте, точности и надежности данных, а также стабильность достижения требуемого результата (далее — валидация данных);
б) утверждает стандартные операционные процедуры использования электронных систем;
в) обеспечивает работу электронных систем таким образом, чтобы при изменении введенных данных вносимые изменения были задокументированы и ранее введенные данные не были удалены;
г) обеспечивает систему защиты данных клинического исследования, предотвращающую несанкционированный доступ к данным, в том числе посредством утверждения списка лиц, имеющих доступ к данным клинического исследования с правом внесения в них изменений и резервного копирования данных;
д) обеспечивает сохранность маскировки клинического исследования, проводимого слепым методом, при вводе и обработке данных в электронной системе.
24. Организатор клинического исследования предоставляет исследователю и медицинской организации протокол клинического исследования и брошюру в текущей редакции до подписания с медицинской организацией договора на проведение клинического исследования и дает время для ознакомления с предоставленной информацией.
25. Организатор клинического исследования должен получить письменное согласие исследователя и уполномоченного лица медицинской организации, посредством подписания протокола или другого документа на:
а) проведение клинического исследования в соответствии с протоколом, настоящими Правилами и требованиями законодательства Российской Федерации в сфере обращения лекарственных средств;
б) соблюдение процедуры регистрации и представления данных клинического исследования;
в) проведение мониторинга и аудита;
г) хранение документов, связанных с проведением клинического исследования, до тех пор, пока организатор клинического исследования не сообщит исследователю и медицинской организации, что данные документы могут быть уничтожены.
26. Организатор клинического исследования обязан в качестве страхователя страховать риск причинения вреда жизни, здоровью пациента в результате проведения клинического исследования лекарственного препарата для медицинского применения за свой счет путем заключения договора обязательного страхования <1>. Условия договора страхования, размер и порядок страховой выплаты устанавливаются в соответствии с постановлением N 714.
<1> С учетом части 1 статьи 44 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
27. При наличии в медицинской организации, в которой планируется проведение клинического исследования, независимого этического комитета, организатор клинического исследования должен получить подтверждение того, что данный независимый этический комитет организован и действует в соответствии с настоящими Правилами и согласие этого независимого этического комитета на проведение клинического исследования.
В случае если согласие независимого этического комитета на проведение клинического исследования выражается посредством внесения изменений в документы и данные клинического исследования, медицинская организация предоставляет организатору клинического исследования копии всех измененных документов и данных.
В случае изменения решения независимого этического комитета относительно проведения клинического исследования, в том числе посредством отзыва ранее данного согласия, медицинская организация незамедлительно уведомляет об этом организатора клинического исследования.
28. Организатор клинического исследования должен иметь данные по безопасности и эффективности исследуемого лекарственного препарата, обосновывающие его применение, а также обновлять брошюру по мере получения в рамках проведения клинического исследования новых данных, влияющих на эффективность и безопасность исследуемого лекарственного препарата.
29. Организатор клинического исследования обеспечивает использование при проведении клинического исследования исследуемых лекарственных препаратов (включая, препараты сравнения и плацебо) произведенных в соответствии с требованиями надлежащей производственной практики, имеющих соответствующие показатели качества и которые в соответствующих случаях закодированы и маркированы в целях обеспечения маскировки.
На первичную упаковку (если для этого существует техническая возможность) и вторичную (потребительскую) упаковку лекарственных препаратов, предназначенных для клинических исследований, должна наноситься надпись: «Для клинических исследований» <1>.
<1> Часть 8 статьи 46 Федерального закона о 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
30. Для клинических исследований, проводимых слепым методом, система кодирования исследуемого лекарственного препарата должна включать в себя механизм, позволяющий в экстренных случаях, быстро идентифицировать данный лекарственный препарат, при этом не допускающий возможности незаметно раскрыть код.
31. Если лекарственная форма исследуемого лекарственного препарата или препарата сравнения была изменена в ходе клинического исследования, то до использования новой лекарственной формы в клиническом исследовании должны быть получены результаты соответствующих дополнительных исследований данной лекарственной формы препарата, необходимые для оценки степени влияния данных изменений на фармакокинетику исследуемого лекарственного препарата, его безопасность и эффективность. В таком случае организатор клинического исследования обеспечивает внесение изменений в протокол.
32. Организатор клинического исследования обеспечивает поступление в медицинскую организацию, в которой проводится клиническое исследование, исследуемого лекарственного препарата.
Организатор клинического исследования должен иметь исследуемый лекарственный препарат в количестве, необходимом для проведения клинического исследования и подтверждения его соответствия требованиям фармакопейных статей либо в случае их отсутствия нормативной документации или нормативного документа, а также вести учет анализов и характеристик образцов лекарственного препарата из партии (серии).
Поступление исследуемого лекарственного препарата в медицинские организации, его возврат, уничтожение исследуемого лекарственного препарата подробно и в срок документируется организатором клинического исследования.
Организатор клинического исследования осуществляет постоянную оценку безопасности исследуемого лекарственного препарата и уведомляет всех занятых в клиническом исследовании исследователей и медицинские организации о полученных данных, которые могут неблагоприятно отразиться на безопасности участников клинического исследования и (или) повлиять на проведение клинического исследования.
33. Организатор клинического исследования сообщает всем участвующим в клиническом исследовании исследователям, медицинским организациям и Федеральной службе по надзору в сфере здравоохранения обо всех серьезных и непредвиденных нежелательных реакциях, а также представляет в Федеральную службу по надзору в сфере здравоохранения периодические отчеты по безопасности исследуемого препарата в установленном порядке <1>.
<1> Приказ Минздравсоцразвития России от 26 августа 2010 г. N 757н «Об утверждении порядка осуществления мониторинга безопасности лекарственных препаратов для медицинского применения, регистрации побочных действий, серьезных нежелательных реакций, непредвиденных нежелательных реакций при применении лекарственных препаратов для медицинского применения» (зарегистрирован Министерством юстиции Российской Федерации 31 августа 2010 г., регистрационный N 18324).
34. Организатор клинического исследования осуществляет мониторинг клинического исследования, включающий деятельность по контролю за ходом клинического исследования, по обеспечению его проведения, сбору данных и представлению результатов в соответствии с протоколом, стандартными операционными процедурами, настоящими Правилами и требованиями законодательства Российской Федерации об обращении лекарственных средств.
Для проведения мониторинга организатор клинического исследования назначает физическое лицо, обладающее научными и (или) специальными знаниями, необходимыми для проведения мониторинга клинического исследования.
Лицо, назначенное организатором клинического исследования для проведения мониторинга, обязано соблюдать стандартные операционные процедуры организатора клинического исследования.
35. Целью мониторинга клинического исследования является установление того, что:
а) права и благополучие участников клинического исследования защищены;
б) полученные в ходе клинического исследования данные являются точными, полными и подтверждаются первичной документацией;
в) клиническое исследование проводится в соответствии с утвержденной текущей версией протокола и изменений к нему, требованиями настоящих Правил и законодательства Российской Федерации об обращении лекарственных средств.
36. Для достижения целей мониторинга клинического исследования лицо, назначенное организатором клинического исследования для проведения мониторинга:
а) обеспечивает взаимодействие между организатором клинического исследования, медицинской организацией и исследователем;
б) проверяет и контролирует наличие квалификации исследователя и ресурсов медицинской организации необходимых для проведения клинического исследования, включая лаборатории, оборудование и персонал;
в) в отношении исследуемого лекарственного препарата проверяет:
сроки и условия хранения, количество в объеме, необходимом для проведения клинического исследования;
получение только теми участниками клинического исследования, которым он назначен, и в дозах, установленных протоколом;
инструкции по правильному применению, хранению и порядку возврата предоставлены участникам клинического исследования;
осуществление исследователем контроля и документального оформления получения, применения, возврата, уничтожения исследуемого лекарственного препарата в медицинской организации;
г) проверяет соблюдение исследователем утвержденного протокола и всех изменений к нему, правильность, полноту, точность и сроки оформления исследователем документов и данных клинического исследования;
д) проверяет получение добровольного письменного согласия каждого участника клинического исследования до начала его участия в клиническом исследовании;
е) обеспечивает наличие у исследователя текущей редакции брошюры, иных документов и материалов, необходимых для проведения клинического исследования;
ж) обеспечивает доведение до исследователей необходимой для проведения клинического исследования информации;
з) проверяет соблюдение исследователем критериев отбора участников клинического исследования;
и) проверяет правильность, полноту и сроки регистрации данных клинического исследования, а также порядок ведения документов клинического исследования;
к) проверяет правильность и полноту регистрации в индивидуальной регистрационной карте данных, предусмотренных протоколом, данных об изменениях дозы и (или) терапии, о выявленных нежелательных явлениях и нежелательных реакциях, о пропущенных участником клинического исследования визитах, о невыполненных анализах и обследованиях. Все случаи исключения и выбывания участников клинического исследования из клинического исследования подлежат регистрации и пояснению в индивидуальной регистрационной карте;
л) сообщает исследователю о любых допущенных в индивидуальной регистрационной карте ошибках, пропусках и неразборчивых записях и прослеживает за тем, чтобы соответствующие исправления, были сделаны, датированы, объяснены (если необходимо) и подписаны исследователем;
м) сообщает исследователю об отклонениях от протокола, стандартных письменных процедур организатора, требований настоящих Правил, законодательства Российской Федерации об обращении лекарственных средств и принимает меры по их устранению и недопущению повторения.
37. Письменные отчеты по мониторингу клинического исследования представляются лицом, назначенным для проведения мониторинга организатору клинического исследования в порядке и сроки, установленные стандартными операционными процедурами организатора.
Отчет по мониторингу клинического исследования должен содержать, в том числе сроки проведения мониторинга, наименование медицинской организации, где проводился мониторинг, данные об исследователях, участниках клинического исследователя, с которыми общалось (взаимодействовало) лицо, назначенное для проведения мониторинга, описание проверки, сведения о выявленных недостатках, выводы по результатам мониторинга и иные сведения и данные, касающиеся мониторинга клинического исследования.
38. Организатором клинического исследования клинического исследования осуществляется независимая от мониторинга оценка соответствия проводимого клинического исследования протоколу, стандартным операционным процедурам организатора, настоящим Правилам и требованиям законодательства Российской Федерации об обращении лекарственных средств (далее — аудит клинического исследования).
Для проведения аудита клинического исследования организатор клинического исследования назначает лиц, независимых от всех субъектов клинического исследования и обладающих квалификацией, подготовкой, опытом для проведения аудита.
План и объем аудита клинического исследования разрабатываются и утверждаются организатором клинического исследования с учетом количества участников клинического исследования, типа и сложности клинического исследования, степени риска для участников клинического исследования и иных обстоятельств.
39. При обнаружении в ходе мониторинга или аудита клинического исследования серьезных и (или) повторяющихся случаев несоблюдения исследователем, медицинской организацией установленных требований к проведению клинического исследования организатор клинического исследования прекращает участие исследователя, медицинской организации в клиническом исследовании и вносит изменения в протокол.
40. В срок, не превышающий пяти рабочих дней со дня завершения, приостановления или прекращения клинического исследования лекарственного препарата для медицинского применения, сообщение об этом направляется организатором клинического исследования в Министерство.
41. При многоцентровых клинических исследованиях организатор клинического исследования обеспечивает:
а) проведение клинического исследования всеми медицинскими организациями, участвующими в многоцентровом клиническом исследовании, в строгом соответствии с протоколом клинического исследования;
б) разработку индивидуальных регистрационных карт, позволяющих собрать требуемые данные из всех медицинских организаций, участвующих в многоцентровом клиническом исследовании;
в) документальное закрепление прав и обязанностей медицинских организаций и исследователей, предоставление указанным лицам протокола, стандартных операционных процедур организатора, инструкции по заполнению индивидуальных регистрационных карт до начала клинического исследования.
IV. Организация работы исследователя
42. Руководитель медицинской организации, которая проводит клиническое исследование, назначает исследователя, ответственного за проведение такого исследования и имеющего лечебную специальность, соответствующую проводимому клиническому исследованию, со стажем работы по программам клинических исследований не менее чем три года и по его предложению назначает соисследователей из числа врачей этой медицинской организации <1> (далее — исследователь).
<1> Пункт 1 статьи 40 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
43. Исследователь, соисследователи должны быть ознакомлены с результатами доклинического исследования исследуемого лекарственного препарата, актуальной версией брошюры, протоколом, иными документами и данными, имеющими отношение к проведению клинического исследования.
44. Исследователь осуществляет отбор участников клинического исследования, обеспечивает оказание медицинской помощи участнику клинического исследования <1>.
<1> Часть 2 статьи 98 Федерального закона от 21 ноября 2011 г. N 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011, N 48, ст. 6724).
45 Исследователь, соисследователи должны знать и соблюдать настоящие Правила и иные требования законодательства Российской Федерации об обращении лекарственных средств. Нарушение настоящих Правил, фальсификация результатов клинического исследования влекут за собой ответственность в соответствии с законодательством Российской Федерации <1>.
<1> Пункт 12 статьи 40 Федерального закона от 12 апреля 2010 г. N 61-ФЗ «Об обращении лекарственных средств».
46. Исследователь должен располагать временем и ресурсами, включая лаборатории, оборудование и персонал, необходимыми для проведения клинического исследования.
47. При наличии согласия участника клинического исследования исследователь сообщает лечащему врачу участника клинического исследования об участии последнего в клиническом исследовании.
48. Исследователь проводит клиническое исследование в соответствии с протоколом.
Исследователь обязан соблюдать протокол, не должен вносить в него изменения без решения Министерства и одобрения независимого этического комитета за исключением случаев, когда требуется устранить непосредственную угрозу жизни и (или) здоровья участника клинического исследования.
Любое отклонение от утвержденного протокола оформляется документально и в кратчайшие сроки направляется для рассмотрения и согласования в независимый этический комитет и организатору клинического исследования для согласования.
49. Исследователь сообщает в независимый этический комитет:
а) об отклонениях от протокола или изменениях протокола, произведенных для устранения угрозы жизни и (или) здоровья участника клинического исследования;
б) об изменениях, непосредственно влияющих на проведение клинического исследования и (или) увеличивающих риск участия в клиническом исследовании;
в) обо всех нежелательных реакциях, которые являются одновременно серьезными и непредвиденными;
г) о новых данных, которые могут свидетельствовать о возрастании риска для участников клинического исследования или могут неблагоприятно повлиять на ход клинического исследования.
50. Исследователь должен обеспечить применение участниками клинического исследования исследуемых лекарственных препаратов в соответствии с протоколом и соблюдать предусмотренную протоколом методику рандомизации (распределения участников клинического исследования по группам лечения или контроля случайным образом, позволяющего свести к минимуму субъективность) и обеспечить раскрытие кода только в соответствии с протоколом.
Если клиническое исследование проводится слепым методом исследователь документально оформляет и объясняет организатору клинического исследования любое преждевременное раскрытие кода исследуемых лекарственных препаратов.
51. Исследователь осуществляет учет исследуемых лекарственных препаратов и препаратов сравнения, в том числе ведет учет их поступлений, фактического наличия, количества использования каждым участником клинического исследования, уничтожения, а также возврата организатору клинического исследования.
Исследователь может передать обязанности по учету исследуемых лекарственных препаратов и препаратов сравнения работнику аптечной организации или иному лицу, подконтрольному исследователю.
Записи по учету должны включать в себя даты, количество, номера партий, серий, сроки годности (где применимо) и уникальные коды исследуемых лекарственных препаратов и препаратов сравнения и участников клинического исследования.
Исследователь должен вести записи, подтверждающие, что участники клинического исследования получали исследуемые лекарственные препараты и (или) препараты сравнения в дозах и количествах, предусмотренных протоколом клинического исследования.
52. Исследователь информирует участника клинического исследования или его законного представителя:
а) о том, что клиническое исследование носит экспериментальный характер, участие лица в клиническом исследовании является добровольным и оно может отказаться от участия в клиническом исследовании в любой момент;
б) о цели клинического исследования, его продолжительности и приблизительном количестве участников;
в) о вариантах лечения в процессе клинического исследования и вероятности случайного распределения в одну из групп лечения;
г) о процедурах клинического исследования, включая все инвазивные процедуры;
д) об обязанностях участника клинического исследования;
е) об ожидаемых риске и (или) пользе для участника клинического исследования, а также, в соответствующих случаях, для эмбриона, плода или грудного ребенка;
ж) об иных, помимо предусмотренных протоколом процедурах или методах лечения, которые могут быть доступны участнику клинического исследования, а также их потенциальных выгоде, пользе, риске;
з) о компенсации и (или) лечении, доступные участнику клинического исследования в случае причинения вреда его здоровью в результате участия в клиническом исследовании;
и) о планируемых выплатах участнику клинического исследования за его участие в клиническом исследовании, если таковые предусмотрены;
к) о планируемых расходах участника клинического исследования, если таковые ожидаются, связанные с его участием в клиническом исследовании;
л) о том, что участник клинического исследования или его законный представитель, подписывая информационный листок пациента, дает разрешение на доступ лицу, назначенному для проведения мониторинга, аудиторов, независимых этических комитетов, уполномоченных органов к медицинским записям участника клинического исследования;
м) о том, что записи, идентифицирующие участника клинического исследования, будут сохранены в тайне, раскрытие их допускается в соответствии с законодательством Российской Федерации и при публикации результатов клинического исследования конфиденциальность данных участника клинического исследования будет сохранена;
н) о том, что участник клинического исследования или его законный представитель будет незамедлительно ознакомлен с новой информацией, способной повлиять на его желание продолжать участие в клиническом исследовании;
о) о лицах, к которым можно обратиться для получения дополнительной информации о клиническом исследовании, и правах участников клинического исследования;
п) о возможных обстоятельствах и (или) причинах, по которым участие лица в клиническом исследовании может быть прекращено.
53. Информация о клиническом исследовании должна содержать как можно меньше специальных терминов и быть понятна участнику клинического исследования или его законному представителю.
54. Исследователь перед получением информированного добровольного согласия должен предоставить участнику клинического исследования, его законному представителю время, необходимое для принятия решения об участии в клиническом исследовании или отказе от такого участия. Участник клинического исследования или его законный представитель вправе получить исчерпывающие и достоверные ответы на все вопросы о клиническом исследовании.
55. Перед включением его в клиническое исследование участник клинического исследования, его законный представитель должен получить подписанный и датированный экземпляр информационного листка пациента и иные материалы, касающиеся проведения клинического исследования. В период проведения клинического исследования участнику клинического исследования сообщается обо всех изменениях в документы и данные клинического исследования, касающиеся его участия в клиническом исследовании.
56. В срок, установленный протоколом, исследователь обязан сообщать организатору клинического исследования обо всех серьезных нежелательных реакциях, за исключением тех, которые в протоколе или в брошюре определены как не требующие немедленного сообщения.
После первого сообщения о серьезных нежелательных реакциях исследователь в кратчайшие сроки представляет организатору клинического исследования подробный письменный отчет. Первый и последующие отчеты должны идентифицировать участников клинического исследования по присвоенным им уникальным кодам.
57. При сообщениях о смерти участника клинического исследования исследователь обязан по запросу организатора клинического исследования, независимого этического комитета, Министерства и (или) Федеральной службы по надзору в сфере здравоохранения предоставить любую дополнительную информацию относительно данного случая, в том числе протокол вскрытия и посмертный эпикриз.
58. В случае возникновения опасности для жизни, здоровья участника клинического исследования, исследователь обязан проинформировать об этом руководителя медицинской организации и организатора клинического исследования лекарственного препарата в течение 24 часов. Решение о приостановлении клинического исследования принимают руководитель медицинской организации и (или) организатор клинического исследования, решение о прекращении такого исследования принимает уполномоченный федеральный орган исполнительной власти на основании сообщения в письменной форме руководителя медицинской организации или организатор клинического исследования.
При досрочном прекращении клинического исследования или его приостановлении исследователь и (или) медицинская организация, в которой проводилось клиническое исследования обязаны незамедлительно информировать участников клинического исследования, обеспечить им необходимое лечение и наблюдение, проинформировать организатора клинического исследования, независимый этический комитет с предоставлением подробного письменного объяснения причин приостановления или прекращения клинического исследования.
59. Исследователь обеспечивает полное и достоверное ведение документов клинического исследования, включая записи на бумажных, электронных, магнитных или оптических носителях, сканограммы, рентгеновские снимки, электрокардиограммы, которые описывают методы, организацию и (или) результаты клинического исследования.
60. Хранение документов клинического исследования осуществляется медицинской организацией в соответствии с условиями договора, заключенного с организатором клинического исследования, или если данное условие не оговорено, как правило, в течение двух лет после государственной регистрации лекарственного препарата в Российской Федерации или официального прекращения клинической разработки исследуемого лекарственного препарата.
61. По завершении клинического исследования исследователь сообщает об этом руководителю медицинской организации, подготавливает отчет в соответствии с требованиями пункта 9 настоящих Правил и представляет его организатору клинического исследования и в независимый этический комитет.
Руководство по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов
(в ред. Решения Коллегии Евразийской экономической комиссии
от 11.10.2022 N 143)
В соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года, статьeй 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года и пунктом 15 перечня актов Евразийской экономической комиссии по вопросам регулирования общих рынков лекарственных средств и медицинских изделий в рамках Евразийского экономического союза на 2017 – 2019 годы (приложение к распоряжению Совета Евразийской экономической комиссии «Об актах Евразийской экономической комиссии по вопросам регулирования общих рынков лекарственных средств и медицинских изделий в рамках Евразийского экономического союза» от 17 мая 2017 г. N 15) Коллегия Евразийской экономической комиссии РЕШИЛА:
Утвердить прилагаемое Руководство по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов.
Настоящее Решение вступает в силу по истечении 6 месяцев с даты его официального опубликования.
При этом исходить из того, что в случае, если реализация положений указанного Руководства осуществляется в соответствии с актами Евразийской экономической комиссии, применение таких положений осуществляется с даты вступления в силу (с даты начала применения) соответствующих актов. соответствующих актов.
Председатель Коллегии
Евразийской экономической комиссии
Т.Саркисян
УТВЕРЖДЕНО
Решением Коллегии
Евразийской экономической комиссии
от 26 ноября 2019 г. N 202
РУКОВОДСТВО
ПО ДОКЛИНИЧЕСКИМ ИССЛЕДОВАНИЯМ БЕЗОПАСНОСТИ В ЦЕЛЯХ
ПРОВЕДЕНИЯ КЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ И РЕГИСТРАЦИИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
(в ред. Решения Коллегии Евразийской экономической комиссии
от 11.10.2022 N 143)
I. Общие положения
1. Целью настоящего Руководства является установление единого порядка проведения доклинических исследований безопасности лекарственных препаратов (далее — доклинические исследования) для их последующей регистрации в рамках Евразийского экономического союза. В настоящем Руководстве отражена взаимосвязь между необходимостью проведения доклинических исследований и проведением клинических исследований с участием человека.
2. Настоящее Руководство направлено на обеспечение:
безопасной, этичной разработки и внедрения новых лекарственных препаратов;
своевременного проведения клинических исследований;
сокращения использования в исследованиях лабораторных животных в соответствии с принципами 3R (замена, улучшение, сокращение) (replacement, refinement, reduction);
сокращения использования других ресурсов при разработке лекарственных препаратов.
Необходимо учитывать возможность использования новых альтернативных in vitro методов оценки безопасности, которые при их надлежащей валидации и одобрении уполномоченными органами (экспертными организациями) государств — членов Евразийского экономического союза (далее соответственно — государства-члены, Союз) могут заменить существующие стандартные методы.
3. Настоящее Руководство составлено в целях гармонизации доклинических исследований, направленных на обоснование проведения клинической разработки лекарственных препаратов на различных ее этапах.
4. Настоящее Руководство содержит указания относительно вида, продолжительности и сроков проведения доклинических исследований в качестве обоснования проведения клинических исследований и регистрации лекарственных препаратов.
5. Доклиническая оценка безопасности лекарственных препаратов в целях их регистрации предусматривает проведение:
а) фармакологических исследований;
б) исследований общетоксических свойств;
в) токсикокинетических и доклинических фармакокинетических исследований;
г) исследований репродуктивной токсичности;
д) исследований генотоксичности;
е) оценки канцерогенного потенциала в отношении лекарственных препаратов, вызывающих особые опасения или предназначенных для длительного применения.
6. Необходимость проведения доклинических исследований в целях оценки фототоксичности, иммунотоксичности, токсичности на неполовозрелых животных и склонности к возникновению лекарственной зависимости определяется в индивидуальном порядке.
7. Настоящее Руководство применяется на этапе разработки лекарственных препаратов и содержит общие требования к процессу доклинической разработки лекарственных препаратов. Планирование и дизайн доклинических исследований и клинических исследований с участием человека основываются на подходе, удовлетворительном с научной и этической точек зрения.
8. Исследования безопасности биотехнологических лекарственных препаратов необходимо проводить в соответствии с требованиями глав 5.3 и 5.4 Правил проведения исследований биологических лекарственных средств Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 89. При проведении исследования безопасности биотехнологических лекарственных препаратов настоящее Руководство применяется только в отношении сроков проведения доклинических исследований в зависимости от фазы клинической разработки биотехнологических лекарственных препаратов.
Результаты исследований безопасности лекарственных препаратов, длительно применяемых в медицинской практике и содержащих действующие вещества, данные о доклиническом исследовании которых ограничены или отсутствуют, допускается представлять в составе смешанных регистрационных досье лекарственных препаратов. Составление документов о доклинических исследованиях для смешанных регистрационных досье лекарственных препаратов осуществляется в соответствии с требованиями, согласно приложению N 1.
9. В целях оптимизации и ускорения разработки лекарственных препаратов для лечения жизнеугрожающих и тяжелых заболеваний (например, распространенного рака, резистентной ВИЧ-инфекции, состояния, обусловленного врожденной ферментативной недостаточностью), не имеющих эффективной терапии, допустим индивидуальный подход к токсикологической оценке и клинической разработке. В этих случаях, а также в отношении лекарственных препаратов, созданных на основе инновационных терапевтических методов (например, малой интерферирующей РНК (siRNA) и адъювантных вакцин), в программе доклинического исследования допустимо сокращение, отказ от проведения или добавление определенных исследований. Для решения вопроса об объеме изменений программы доклинического исследования необходимо руководствоваться положениями актов органов Союза в сфере обращения лекарственных средств или законодательством государств-членов.
10. Доклинические исследования необходимо проводить с учетом требований пунктов 34 и 35 Правил регистрации и экспертизы лекарственных средств для медицинского применения, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78 (далее — Правила регистрации и экспертизы).
11. Доклинические исследования, проведенные за пределами таможенной территории Союза, должны быть выполнены с учетом указаний пункта 35 Правил регистрации и экспертизы, а также соответствовать настоящему Руководству и другим актам органов Союза в сфере обращения лекарственных средств.
12. По вопросам, не урегулированным в настоящем Руководстве, заявители вправе обратиться за научной консультацией в уполномоченные органы (экспертные организации) государств-членов в соответствии с пунктом 26 Правил регистрации и экспертизы.
13. Разработка лекарственного препарата — поэтапный процесс, предусматривающий оценку данных о его эффективности и безопасности для животных и человека. К целям доклинической оценки безопасности относится установление:
характеристик токсического действия в отношении органов-мишеней;
зависимости токсического действия от дозы;
зависимости токсического действия от экспозиции;
потенциальной обратимости токсического действия (в соответствующих случаях).
Полученные сведения используют для оценки начальной безопасной дозы и диапазона доз в исследованиях с участием человека, а также для определения соотношения «польза — риск» и параметров клинического мониторинга потенциальных нежелательных эффектов.
14. Доклинические исследования, несмотря на ограниченный характер на начальном этапе клинической разработки, должны быть достаточными для описания потенциальных нежелательных реакций, которые могут возникнуть в условиях обоснованного клинического исследования.
15. Клинические исследования проводятся в целях изучения эффективности и безопасности лекарственного препарата, начиная с относительно низкой системной экспозиции у небольшого числа субъектов. За ними следуют клинические исследования, в которых экспозиция лекарственного препарата увеличивается, как правило, за счет продолжительности применения и (или) размера популяции пациентов.
16. Клинические исследования необходимо расширять, если получены результаты, подтверждающие достаточную безопасность лекарственного препарата в ранее проведенном клиническом исследовании (исследованиях), а также при наличии дополнительных данных доклинических исследований безопасности, которые становятся доступными по мере выполнения клинической разработки.
17. Серьезные нежелательные эффекты, выявленные на клиническом и (или) доклиническом этапах разработки лекарственного препарата, могут повлиять на продолжение клинической разработки. В целях определения целесообразности разработки дизайна и проведения дополнительных доклинических и (или) клинических исследований эти данные необходимо рассматривать в рамках общего плана клинической разработки.
18. Клинические исследования проводятся с разделением по фазам клинической разработки, каждая из которых имеет различные цели. Поскольку допускается объединение фаз клинической разработки, в настоящем Руководстве требования к доклиническим исследованиям соотнесены не только с фазами клинической разработки, но и с характером клинических исследований и целевой популяцией.
19. Понятия, используемые в настоящем Руководстве, применяются в значениях, определенных в Правилах надлежащей клинической практики Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 79.
II. Выбор максимальных доз в исследованиях
общей токсичности
20. В случае если в токсикологических исследованиях удается охарактеризовать все потенциальные клинически значимые эффекты при введении диапазона доз лекарственного препарата ниже, чем его максимальная переносимая доза (MTD), то устанавливать максимальную переносимую дозу в каждом таком исследовании не требуется.
21. К другим подходящим предельным дозам относятся дозы, создающие в организме высокие кратные экспозиции или насыщение экспозиции, либо максимальная достижимая доза (MFD). Такие предельные дозы позволяют не использовать в отношении животных дозы, которые не дают дополнительных сведений для прогнозирования клинической безопасности. Подход к выбору максимальных доз в исследованиях общей токсичности приведен на рисунке. Этот подход согласуется с указаниями в отношении дизайна исследований репродуктивной токсичности и канцерогенности, в которых уже определены предельные дозы и (или) экспозиции.
Рисунок. Подход к выбору максимальных доз в исследованиях
общей токсичности
22. Предельные дозы для исследований токсичности при однократном и повторном (многократном) введении, равные 1 000 мг/кг/сут. для грызунов и негрызунов, признаны приемлемыми во всех случаях, за исключением случаев, если:
доза 1 000 мг/кг/сут. не обеспечивает 10-кратного превышения клинической экспозиции, а клиническая доза превышает 1 г/сут. В этом случае дозы в токсикологических исследованиях необходимо ограничить 10-кратным превышением экспозиции, либо дозой, равной 2 000 мг/кг/сут., либо максимальной достижимой дозой в зависимости от того, какая из этих доз меньше;
доза 2 000 мг/кг/сут. меньше клинической экспозиции. В этом случае может потребоваться увеличение дозы вплоть до максимальной достижимой дозы.
23. Дозы, обеспечивающие 50-кратное превышение средней экспозицией клинической экспозиции, как правило, также приемлемы в качестве максимальной дозы в исследованиях токсичности при однократном введении и токсичности с повторным (многократным) введением всем видам животных при условии, что установлена линейная зависимость «доза — концентрация» исходного соединения или фармакологически активной молекулы пролекарства в биологическом материале.
Экспозицию необходимо определять для исходного соединения или фармакологически активной молекулы пролекарства по групповому среднему значению площади под кривой «плазменная концентрация — время» (AUC) с момента приема лекарственного препарата. В некоторых случаях превышение экспозиции допускается определять по групповым средним значениям максимальной плазменной концентрации (Cmax) (например, если соединение или класс соединений способны вызывать острые сердечно-сосудистые изменения или клинические симптомы, связанные с воздействием на центральную нервную систему).
24. Для обоснования проведения клинических исследований III фазы необходимо, как правило, определить дозолимитирующую токсичность не менее чем на 1 виде животных с предельной дозой, обеспечивающей 50-кратное превышение экспозиции. В противном случае рекомендуется провести исследование продолжительностью 1 месяц (или более), на одном виде животных с предельной дозой, равной 1 000 мг/кг, максимальной достижимой дозе или максимальной переносимой дозе, с использованием меньшей из этих доз. Такое исследование может не потребоваться, если в исследовании меньшей продолжительности установлена дозолимитирующая токсичность при дозах, превышающих дозы, обеспечивающие 50-кратное превышение экспозиции.
25. Если в исследование общей токсичности включаются конечные точки генотоксичности, то подходящую максимальную дозу необходимо выбирать на основе максимальной достижимой дозы, максимальной переносимой дозы или предельной дозы, равной 1 000 мг/кг/сут.
III. Исследования фармакологической безопасности
26. Исследования фармакологической безопасности и фармакодинамические исследования проводятся в соответствии с руководством по исследованию фармакологической безопасности лекарственных препаратов для медицинского применения, принимаемым Евразийской экономической комиссией.
27. Основной набор (батарея) исследований фармакологической безопасности включает в себя оценку воздействия на сердечно-сосудистую, центральную нервную и дыхательную системы, которая должна проводиться до начала клинической разработки в соответствии с руководством по исследованию фармакологической безопасности лекарственных препаратов для медицинского применения.
28. Вспомогательные и последующие исследования фармакологической безопасности при необходимости допускается проводить на более поздних этапах клинической разработки.
29. В целях уменьшения использования животных в исследованиях фармакологической безопасности необходимо предусмотреть возможность замены этих исследований оценкой определенных конечных точек в рамках исследований общей токсичности in vivo.
30. Исследования первичной фармакодинамики (in vivo и (или) in vitro) направлены на изучение механизма действия и (или) эффектов действующего вещества по отношению к его целевой терапевтической мишени. Такие исследования проводятся в начальной фазе фармацевтической разработки лекарственного препарата, их проведение допускается без учета требований Правил надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81 (далее — Правила лабораторной практики). Результаты этих исследований учитываются при обосновании выбора дозы (режима дозирования) в доклинических и клинических исследованиях.
IV. Токсикокинетические и фармакокинетические исследования
31. До начала клинических исследований необходимо оценить метаболический профиль и степень связывания с белками плазмы у животных и человека in vitro, а также проанализировать данные о системной экспозиции, полученные в исследованиях на видах животных, использованных для изучения токсичности при повторном (многократном) введении действующего вещества лекарственного препарата, в соответствии с руководством по изучению токсикокинетики и оценки системного воздействия в токсикологических исследованиях, принимаемом Евразийской экономической комиссией.
32. До назначения лекарственного препарата большому числу субъектов или в течение длительного времени (как правило, до начала III фазы клинических исследований лекарственных препаратов) необходимо располагать более подробными данными о фармакокинетике (включая сведения об абсорбции, распределении, метаболизме и выведении) у испытуемых видов животных и in vitro биохимическими данными, значимыми для выявления потенциальных лекарственных взаимодействий. Эти данные используются для сравнения метаболитов человека и животных, а также для определения необходимости проведения дополнительных исследований.
33. Доклиническое установление характеристик метаболита (метаболитов) человека необходимо осуществлять только в том случае, если его экспозиция превышает 10% от суммарной экспозиции лекарственного препарата и величина экспозиции у человека значимо превышает экспозицию, наблюдавшуюся в токсикологических исследованиях. Такие исследования проводятся для обоснования проведения клинических исследований III фазы. В отношении лекарственных препаратов, суточная доза которых менее 10 мг, допускается обосновывать более высокий предел экспозиции для проведения доклинического исследования метаболита с уровнем, превышающим 10% от суммарной экспозиции лекарственного препарата. Некоторые метаболиты (например, большинство глутатион-конъюгатов) не представляют токсикологической опасности и не требуют проведения исследований. Доклиническое установление характеристик метаболитов с выявленной причиной для опасений (например, метаболита, свойственного только человеку) рассматривается в индивидуальном порядке.
V. Исследования токсичности при однократном введении
лекарственного препарата
34. Данные по токсичности при однократном введении лекарственного препарата традиционно получают в исследованиях токсичности с введением млекопитающим двух видов с использованием парентерального пути введения и пути введения, указанного в проекте общей характеристики лекарственного препарата. Вместе с тем эти данные также можно получить в надлежащим образом проводимых исследованиях эскалации дозы или краткосрочных исследованиях по подбору диапазона доз, которые позволяют определить максимальную переносимую дозу у животных, используемых в исследованиях общей токсичности.
35. При наличии данных токсичности при однократном введении лекарственного препарата, полученных при проведении других исследований, отдельные исследования с однократным введением лекарственного препарата проводить не требуется. Если введение лекарственного препарата в клинических условиях обосновано соответствующими исследованиями токсичности при повторном (многократном) введении лекарственного препарата в соответствии с Правилами лабораторной практики, то исследования токсичности при однократном введении лекарственного препарата допустимо ограничить только исследованиями токсичности при данном клиническом способе введения лекарственного препарата и исследованиями токсичности при однократном введении лекарственного препарата, проведенными без учета требований Правил лабораторной практики. Летальность не должна являться целевой конечной точкой в исследованиях токсичности при однократном введении лекарственного препарата.
36. В некоторых случаях (например, при исследовании микродоз) исследования токсичности при однократном введении лекарственного препарата или исследования однократного введения лекарственного препарата могут быть главным обоснованием проведения исследований с участием человека. В этих случаях выбор максимальной дозы может отличаться от описанного в разделе II настоящего Руководства, но он должен соответствовать планируемой клинической дозе и пути введения лекарственного препарата. Эти исследования необходимо проводить в соответствии с Правилами лабораторной практики.
37. Данные токсичности при однократном введении лекарственного препарата могут использоваться в целях прогнозирования последствий передозировки у человека и должны быть доступны для обоснования проведения III фазы клинических исследований. При изучении показаний к применению лекарственного препарата у популяций пациентов, имеющих высокий риск передозировки (например, при депрессии, боли, деменции), в клинических исследованиях, проводимых в амбулаторных условиях, может потребоваться более ранняя оценка токсичности при однократном введении лекарственного препарата.
VI. Исследования токсичности при повторном (многократном)
введении лекарственного препарата
38. Рекомендуемая продолжительность исследования токсичности при повторном (многократном) введении лекарственного препарата, как правило, зависит от продолжительности, показаний к применению и направленности планируемого клинического исследования. Продолжительность токсикологических исследований на животных, проводимых на млекопитающих 2 видов (один из которых относится к негрызунам), должна быть равной или превышать продолжительность клинических исследований с участием человека вплоть до максимальной рекомендованной продолжительности исследований токсичности при повторном (многократном) введении лекарственного препарата (в соответствии с требованиями согласно приложению N 2). Предельные дозы (экспозиции), подходящие для исследований токсичности при повторном (многократном) введении лекарственного препарата, описаны в разделе II настоящего Руководства.
1. Исследования клинической разработки
39. Исследования токсичности при повторном (многократном) введении лекарственного препарата животным двух видов (один из которых относится к негрызунам) с минимальной продолжительностью 2 недели, предусмотренной приложением N 2 к настоящему Руководству, как правило, достаточны для обоснования проведения каждого исследования клинической разработки продолжительностью менее 2 недель.
40. Проведение исследований клинической разработки большей продолжительности необходимо обосновать результатами исследований токсичности при повторном (многократном) введении лекарственного препарата, по меньшей мере эквивалентной продолжительности. Исследования на негрызунах в течение 6 и 9 месяцев применения лекарственного препарата также используются для обоснования дозирования в клинических исследованиях, превышающих 6 месяцев, за исключением случаев, указанных в приложении N 2 к настоящему Руководству.
2. Исследования для обоснования регистрации
лекарственного препарата
41. Для регистрации лекарственного препарата необходима бльшая продолжительность доклинических исследований в связи с увеличением целевой популяции пациентов, которые будут подвергаться риску, связанному с приемом лекарственного препарата, а также менее контролируемыми условиями применения препарата по сравнению с клиническими исследованиями. Продолжительность исследований токсичности при повторном (многократном) введении лекарственного препарата, необходимых для обоснования терапии различной продолжительности, определяется в соответствии с приложением N 3. Вместе с тем при небольшом числе состояний с рекомендуемой продолжительностью применения лекарственного препарата от 2 недель до 3 месяцев, но при наличии большого клинического опыта, подтверждающего более широкое и длительное применение лекарственного препарата сверх рекомендуемой продолжительности применения (например, при тревоге, сезонном аллергическом рините, боли), продолжительность исследований больше соответствует случаям, когда рекомендуемая продолжительность применения лекарственного препарата превышает 3 месяца.
VII. Доклинические исследования, проводимые в целях
определения величины первой дозы у человека
42. Определение величины начальной дозы для человека — важный элемент обеспечения безопасности субъектов, участвующих в исследованиях, впервые проводимых с участием человека.
43. При определении рекомендованной начальной дозы лекарственного препарата для человека необходимо учитывать все актуальные данные доклинических исследований, включающие фармакологическую зависимость «доза — эффект», фармакологический (токсикологический) профиль и фармакокинетику.
44. При определении рекомендованной начальной дозы наиболее значимой является величина дозы, не оказывающей явного нежелательного эффекта (NOAEL), установленная в доклинических исследованиях безопасности на подходящих видах животных. Планируемая клиническая начальная доза также может зависеть от разных факторов, включая фармакодинамику, определенные свойства молекулы и дизайн клинических исследований.
45. Поисковые клинические исследования с участием человека могут быть начаты при меньшем объеме данных доклинических исследований, чем объем, который необходим для проведения исследований клинической разработки, в связи с чем величина клинической начальной (и максимальной) дозы может отличаться от величины дозы, определенной на основании полного объема данных доклинических исследований. Критерии выбора начальных доз в различных поисковых клинических исследованиях определяются в соответствии с требованиями согласно приложению N 4.
VIII. Доклинические исследования, проводимые в целях
обоснования проведения поисковых клинических исследований
46. Оптимизированные поисковые клинические исследования и ранний доступ к данным, полученным в исследованиях с участием человека, могут способствовать:
а) правильной оценке изменений физиологических функций в организме человека под влиянием лекарственного препарата (фармакологических свойств лекарственного препарата);
б) установлению характеристик исследуемого лекарственного препарата;
в) оценке соответствия терапевтических мишеней лекарственного препарата заболеванию, для лечения которого он предназначен.
47. Для целей настоящего Руководства под поисковыми клиническими исследованиями понимаются исследования, проводимые на раннем этапе I фазы клинических исследований, предусматривающие ограниченную экспозицию у человека и не предполагающие проведение оценки терапевтической эффективности и клинической переносимости. Эти исследования проводят для изучения таких параметров, как:
фармакодинамические;
фармакокинетические;
биомаркеры, которые могут включать в себя рецепторное связывание и вытеснение лекарственного препарата, изучены по результатам позитронно-эмиссионной томографии (далее — ПЭТ);
другие диагностические параметры.
Субъектами этих исследований могут являться как пациенты из целевой популяции, так и здоровые добровольцы.
48. Объем и вид обосновывающих данных доклинических исследований, необходимых для планирования поисковых клинических исследований, будут зависеть от величины экспозиции у человека с точки зрения используемой максимальной клинической дозы, продолжительности курса лечения и кратности введения. В настоящем разделе и приложении N 4 к настоящему Руководству представлены 5 видов клинических поисковых подходов с программами доклинических исследований, которые разрешены для этих подходов, а также рекомендуемыми начальными дозами и максимальными дозами для этих 5 подходов.
49. Допустимо использование альтернативных подходов, не описанных в настоящем Руководстве (в том числе стратегии разработки биотехнологических препаратов). Такие альтернативные подходы должны быть согласованы с уполномоченным органом (экспертной организацией) государства-члена. Применение альтернативных подходов может уменьшить использование животных при разработке лекарственных препаратов.
50. Во всех случаях для обоснования выбора доз в исследованиях с участием человека необходимо устанавливать характеристики фармакодинамики и фармакологических свойств действующего вещества лекарственного препарата с использованием моделей in vivo и (или) in vitro, приведенных в разделе III и приложении N 4 к настоящему Руководству.
1. Исследования микродоз
51. При исследовании микродоз используются 2 основных поисковых клинических подхода.
Первый подход предполагает применение суммарной дозы, не превышающей 100 мкг, которая может быть введена субъекту в виде однократной дозы или разделенной на несколько доз. Это может способствовать изучению связывания с рецептором-мишенью или распределения в тканях с помощью ПЭТ. Другой целью может служить изучение фармакокинетики с помощью использования меченного изотопом действующего вещества лекарственного препарата или без него.
Второй подход предусматривает не более 5 введений доз с максимальной величиной 100 мкг на введение (суммарно до 500 мкг) субъекту. Этот подход можно использовать для решения задач, аналогичных задачам в первом подходе, но для лигандов, менее активных при ПЭТ. Разрешается проводить клиническое исследование микродоз с внутривенным введением лекарственного препарата, предназначенного для приема внутрь, в отношении которого уже проведен ряд токсикологических исследований с пероральным введением. В этом случае вводимая внутривенно микродоза обосновывается имеющимися данными исследований пероральной токсичности, описанными в приложениях N 2 и N 4 (подход 3-й) к настоящему Руководству, если достигнуто достаточное превышение экспозиции. В таком случае местная переносимость лекарственного препарата при внутривенном введении не изучается, поскольку вводимая доза слишком мала (до 100 мкг). При использовании нового внутривенного носителя необходимо оценить его местную переносимость.
2. Клинические исследования с однократным
введением субтерапевтических доз или доз предполагаемого
терапевтического диапазона
52. Третий поисковый клинический подход предусматривает проведение клинического исследования обычно начинающегося с однократного введения субтерапевтических доз с возможным повышением до фармакологического или предполагаемого терапевтического диапазона доз предусмотренного приложением N 4 к настоящему Руководству. Введение максимальной допустимой дозы лекарственного препарата должно основываться на данных доклинических исследований. В дальнейшем на основании клинических данных, полученных в ходе данного исследования, она может быть уменьшена. Такой подход позволяет:
определить фармакокинетические параметры радиоактивно не меченного лекарственного препарата при условии его введения в прогнозируемой фармакодинамически активной дозе или в близком к ней диапазоне доз;
дать оценку участия мишени или фармакологических свойств лекарственного препарата после введения однократной дозы.
Этот подход не предназначен для обоснования определения максимальной переносимой клинической дозы.
53. В рамках расширенного исследования токсичности при однократном введении лекарственного препарата необходимо, как правило, оценить данные по гематологии, биохимии, некропсии и гистопатологии (только для контроля и максимальной дозы, если при максимальной дозе не обнаруживается патология, обусловленная применением препарата) после однократного введения лекарственного препарата с последующим изучением спустя 2 недели для оценки отсроченной токсичности и (или) восстановления. Стандартный дизайн исследования на грызунах предусматривает использование 10 особей каждого пола на группу, оцениваемых на следующий день после введения, и 5 особей каждого пола для дозы (доз), оцениваемой на 14-й день после введения лекарственного препарата. Стандартный дизайн исследований на негрызунах предусматривает использование 3 особей каждого пола во всех группах для оценки (изучения) данных по фармакологической безопасности на 2-й день и 2 особей каждого пола для дозы (доз), оцениваемой на 14-й день. Проведение клинических исследований продолжительностью менее 14 дней разрешается обосновывать исследованиями токсичности с той же продолжительностью.
3. Клинические исследования с повторным (многократным)
введением лекарственного препарата
54. Для клинических исследований с повторным (многократным) введением лекарственного препарата четвертый и пятый поисковые клинические подходы совместно с программами доклинических исследований приведены в приложении N 4 к настоящему Руководству. Указанные подходы позволяют обосновать продолжительность дозирования до 14 дней в целях изучения фармакокинетики и фармакодинамики у человека в терапевтическом диапазоне, но не предназначены для обоснования максимальной переносимой клинической дозы.
55. Четвертый подход предусматривает проведение 2-недельных исследований токсичности при повторном (многократном) введении лекарственного препарата грызунам и негрызунам, в которых выбор доз основан на экспозиции, кратной ожидаемому групповому среднему значению площади под кривой «плазменная концентрация — время» при максимальной клинической дозе.
56. Пятый подход предусматривает проведение 2-недельных исследований токсичности при повторном (многократном) введении лекарственного препарата грызунам и подтверждающих исследований на негрызунах, направленных на подтверждение того, что доза, не оказывающая явного нежелательного эффекта (NOAEL), установленная для грызунов, также не является токсичной дозой для негрызунов. Если при применении дозы, создающей экспозицию, равную дозе, не оказывающей явного нежелательного эффекта (NOAEL-экспозиция) для грызунов, токсическое воздействие наблюдается у негрызунов, то клиническое применение лекарственного препарата необходимо отложить до проведения дополнительных доклинических исследований на данном виде негрызунов (как правило, стандартного токсикологического исследования).
IX. Исследования местной переносимости
лекарственного препарата
57. Если местная переносимость лекарственного препарата при его планируемом пути введения изучена во время исследований общей токсичности, выполнение отдельного исследования местной переносимости лекарственного препарата не требуется.
58. Для обоснования ограниченного введения лекарственного препарата с помощью нетерапевтических путей (например, однократного внутривенного введения в целях определения относительной биодоступности лекарственного препарата для приема внутрь) необходимо выполнить исследование местной переносимости при однократном введении одному виду животных. Если ожидаемая системная экспозиция (AUC и Cmax) при нетерапевтическом пути введения перекрывается имеющимися токсикологическими данными, конечными точками исследования местной переносимости являются клинические признаки, макроскопические и микроскопические изменения при гистологическом исследовании места введения лекарственного препарата. Состав лекарственного препарата, предназначенный для изучения местной переносимости, может полностью не совпадать с клиническим составом лекарственного препарата, но должен быть сходным с ним.
59. В случае проведения исследования микродоз с применением внутривенного введения лекарственного препарата, обоснованного токсикологическими данными при пероральном введении, оценка местной переносимости действующего вещества не требуется. При использовании лекарственного препарата с новым составом вспомогательных веществ для внутривенного введения действующего вещества необходимо оценить местную переносимость такого лекарственного препарата.
60. Для парентерального лекарственного препарата до того, как экспозиции данного лекарственного препарата подвергнется большое количество пациентов в клинических исследованиях (например, в клинических исследованиях III фазы), необходимо оценить местную переносимость этого лекарственного препарата в доклинических исследованиях с использованием для его введения мест инъекций, которые не предусмотрены предполагаемым клиническим применением лекарственного препарата. Необходимо проводить однократное паравенозное введение в отношении лекарственных препаратов с внутривенным способом введения. Иные парентеральные пути введения лекарственного препарата необходимо оценивать в индивидуальном порядке.
X. Клинические исследования генотоксичности
лекарственного препарата
61. Проведение доклинических исследований на генные мутации достаточно для обоснования проведения всех клинических исследований с однократным введением лекарственного препарата. В целях обоснования проведения клинических исследований с повторным (многократным) введением лекарственного препарата необходимо провести дополнительную оценку, позволяющую выявить хромосомные повреждения у млекопитающих. Полный набор (батарею) исследований на генотоксичность необходимо завершить до начала II фазы клинических исследований.
62. Если результаты исследований генотоксичности положительны, в целях установления возможности продолжения введения лекарственного препарата человеку, необходимо провести оценку этих результатов, а также при необходимости дополнительные исследования.
63. Доклинические исследования генотоксичности, необходимые для обоснования подходов к проведению поисковых клинических исследований, указаны в разделе VIII настоящего Руководства.
XI. Исследования канцерогенности лекарственного препарата
64. Условия, требующие исследования канцерогенного потенциала лекарственного препарата, установлены в Руководстве по оценке и контролю ДНК-реактивных (мутагенных) примесей в лекарственных средствах и установлению границ потенциального канцерогенного риска (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 6 августа 2019 г. N 23).
65. Если проведение исследований канцерогенности лекарственного препарата требуется для клинического показания, такие исследования канцерогенности следует провести для обоснования регистрации лекарственного препарата с применением по данному показанию. При наличии опасений относительно возможного канцерогенного риска лекарственного препарата результаты исследований канцерогенности необходимо представить до проведения клинических исследований такого лекарственного препарата. Большая продолжительность клинического исследования не рассматривается в качестве существенной причины возможного канцерогенного риска.
66. Исследования канцерогенности лекарственных препаратов, разрабатываемых для лечения тяжелых заболеваний у взрослых или детей, при необходимости разрешается проводить после регистрации лекарственного препарата.
XII. Исследования репродуктивной и онтогенетической
токсичности лекарственного препарата
67. Исследования репродуктивной и онтогенетической токсичности лекарственного препарата необходимо проводить с учетом особенностей популяции, которая будет подвергаться экспозиции этого лекарственного препарата.
1. Мужчины
68. Поскольку оценка репродуктивной системы самцов проводится в рамках проведения исследований токсичности с повторным (многократным) введением лекарственного препарата, разрешается включать мужчин в клинические исследования I и II фаз до проведения специальных исследований фертильности у самцов. Оценка репродуктивной системы у самцов и самок по результатам стандартного гистопатологического исследования яичек и яичников в исследованиях токсичности с повторным (многократным) введением лекарственного препарата (как правило, у грызунов) не менее 2-недельной продолжительности считается сопоставимой по чувствительности к выявлению токсического действия на репродуктивные органы животных со специальными исследованиями фертильности.
69. Исследования фертильности у самцов необходимо провести до начала крупных или длительных клинических исследований (например, клинические исследования III фазы).
2. Женщины без детородного потенциала
70. Если проведены исследования токсичности при повторном (многократном) введении лекарственного препарата, предусматривающие оценку репродуктивных органов у самок, разрешается включать в клинические исследования женщин без детородного потенциала (то есть стерилизованных или находящихся в постменопаузе) без проведения исследований репродуктивной токсичности. При этом постменопауза определяется как отсутствие менструаций в течение 12 месяцев, не обусловленное иными медицинскими причинами.
3. Женщины с детородным потенциалом
71. В отношении женщин с детородным потенциалом существует высокая степень опасности подвергнуть эмбрион или плод непреднамеренной экспозиции лекарственного препарата до получения сведений о потенциальной пользе и потенциальных рисках этого лекарственного препарата.
72. При включении в клинические исследования женщин с детородным потенциалом необходимо описать и минимизировать риск непреднамеренной экспозиции лекарственного препарата у эмбриона или плода. Первый подход к достижению этой цели заключается в проведении исследований репродуктивной токсичности для оценки риска, связанного с самим лекарственным препаратом, и принятия соответствующих мер предосторожности у женщин с детородным потенциалом в клинических исследованиях. Второй подход заключается в ограничении риска за счет принятия мер в целях предотвращения наступления беременности в ходе проведения клинических исследований. К таким мерам относятся:
тесты на беременность (например, по определению свободной -субъединицы хорионического гонадотропина человека);
использование высоконадежных методов контрацепции, которыми считаются как изолированные, так и комбинированные методы, обеспечивающие низкую частоту неудач (то есть менее 1% в год) при их постоянном и правильном применении. При применении метода гормональной контрацепции необходимо учитывать информацию об исследуемом лекарственном препарате и его потенциальном влиянии на контрацепцию;
включение в исследование только после подтверждения наличия менструаций.
Тесты на беременность, выполняемые в ходе проведения исследования, и обучение пациентов мерам предотвращения наступления беременности и контроля наступления беременности должны быть достаточными, чтобы обеспечить приверженность пациентов таким мерам в период экспозиции лекарственного препарата (который может превышать продолжительность исследования).
73. Для дополнительной минимизации риска непреднамеренной экспозиции лекарственного препарата в рамках подходов, описанных в пункте 72 настоящего Руководства, информированное согласие должно основываться на всей имеющейся информации о репродуктивной токсичности лекарственного препарата (например, общей оценке потенциальной токсичности лекарственных препаратов, имеющих родственные структуры и фармакологические эффекты). В случае отсутствия актуальных сведений о репродуктивной токсичности лекарственного препарата в информированном согласии необходимо указать сведения о потенциальных невыявленных рисках для эмбриона или плода.
74. Допускается включать женщин с детородным потенциалом в ранние фазы клинических исследований без доклинического изучения онтогенетической токсичности (исследования эмбриофетального развития) в следующих случаях:
а) интенсивный контроль риска в краткосрочных (например, 2-недельных) клинических исследованиях;
б) преобладание заболевания среди женщин и невозможность достижения цели исследования без включения в клинические исследования женщин с детородным потенциалом, а также принятие достаточных мер для предотвращения наступления беременности.
75. Дополнительными факторами проведения исследований у женщин с детородным потенциалом без проведения доклинических исследований онтогенетической токсичности являются сведения о механизме действия лекарственного препарата (его метаболита), его свойствах, степени экспозиции лекарственного препарата у эмбриона или плода и затруднениях при проведении исследований онтогенетической токсичности на подходящей модели животных (например, исследования онтогенетической токсичности моноклональных антител, для которых по имеющимся научным данным установлена низкая эмбриофетальная экспозиция в период органогенеза, разрешается проводить во время III фазы клинических исследований). При этом исследования онтогенетической токсичности должны быть проведены к моменту подачи регистрационного досье лекарственного препарата на регистрацию и в регистрационное досье должен быть включен полный отчет о данном виде токсичности лекарственного препарата.
76. До проведения основных исследований репродуктивной токсичности разрешается включение в клинические исследования женщин с детородным потенциалом (до 150 человек), которые будут получать исследуемый лекарственный препарат в течение относительно короткого периода (до 3 месяцев), при условии наличия предварительных данных о репродуктивной токсичности лекарственного препарата, полученных на 2 видах животных, и при принятии мер для предотвращения наступления беременности у субъектов исследования. Для достижения этой цели проводится предварительное исследование эмбриофетального развития при введении лекарственного препарата в достаточных дозах. Исследование должно быть проведено с участием не менее 6 самок на группу животных и самок, получавших препарат в период органогенеза. Исследование должно включать в себя оценку выживаемости плода, массы тела, внешнее изучение плода, а также изучение внутренних органов плода. Предварительное доклиническое исследование проводится в соответствии с Правилами лабораторной практики и научными стандартами в области биомедицинских исследований при условии обеспечения прямого доступа заинтересованных лиц к собранным данным.
77. Разрешение на проведение клинических исследований с участием женщин с детородным потенциалом связано с:
а) очень низкой частотой наступления беременности во время контролируемых клинических исследований с участием небольших когорт (до 150 человек);
б) короткой продолжительностью таких исследований. Частота наступления беременности у женщин, впервые предпринимающих попытку забеременеть, составляет примерно 17% на менструальный цикл. Частота наступления беременности в III фазе клинических исследований, проведенных у женщин с детородным потенциалом, составляла менее 0,1% на менструальный цикл, если в ходе этих исследований субъектам рекомендовали не допускать наступления беременности и были предприняты меры для предотвращения наступления беременности. Частота наступления беременности во II фазе клинических исследований ниже, чем в III фазе исследований, но ввиду ограниченного числа включенных в данную фазу исследований женщин величину снижения частоты таких случаев установить невозможно. Основываясь на указанных данных, частота наступления беременности во II фазе исследований, включающих в себя до 150 женщин с детородным потенциалом и продолжительностью до 3 месяцев, должна составлять менее чем 0,5 случая беременности на каждый исследуемый лекарственный препарат;
в) способностью надлежащим образом спланированных предварительных исследований выявлять большинство онтогенетических токсических эффектов, которые могли бы повлиять на включение в клинические исследования женщин с детородным потенциалом.
78. На допустимое к включению в клинические исследование число женщин с детородным потенциалом и продолжительность исследования влияют характеристики популяции, связанные с вероятностью наступления беременности (например, возраст, заболевание и т.д.). Необходимо завершить основные доклинические исследования онтогенетической токсичности до включения в клиническое исследование женщин с детородным потенциалом, за исключением ситуаций, указанных в пунктах 72 — 76 настоящего Руководства.
79. Поскольку оценка репродуктивных органов самок проводится в рамках проведения исследований токсичности при повторном (многократном) введении лекарственного препарата, разрешается включать женщин с детородным потенциалом в клинические исследования I и II фаз с повторным (многократным) введением до проведения исследования фертильности у самок с учетом пункта 68 настоящего Руководства. Для обоснования включения женщин с детородным потенциалом в крупные или продолжительные клинические исследования (например, исследования III фазы) необходимо провести доклинические исследования, направленные на оценку фертильности у самок.
80. В регистрационное досье лекарственного препарата необходимо включить результаты исследований пренатального и постнатального онтогенетического развития потомства животных, подвергшихся экспозиции лекарственного препарата.
81. До включения женщин с детородным потенциалом в клинические исследования, в которых не используются высокоэффективные методы контрацепции, или при неизвестном гестационном статусе необходимо провести полное исследование репродуктивной токсичности у самок и стандартный (полный) набор (батарею) исследований генотоксичности. Высоконадежными методами контрацепции считаются как изолированные, так и комбинированные методы, обеспечивающие низкую частоту неудач (то есть менее 1% в год), при их постоянном и правильном применении. При применении метода гормональной контрацепции необходимо учитывать информацию об исследуемом лекарственном препарате и его потенциальном влиянии на эффективность этого метода гормональной контрацепции.
4. Беременные женщины
82. До включения беременных женщин в клинические исследования необходимо провести исследования репродуктивной токсичности у самок и стандартный (полный) набор (батарею) исследований генотоксичности, а также необходимо оценить имеющиеся данные о безопасности экспозиции изучаемого лекарственного препарата у человека.
XIII. Клинические исследования с участием детей
83. При включении в клинические исследования детей наиболее значимыми сведениями являются данные по безопасности, которые получены с участием взрослых субъектов, которые, как правило, должны быть доступны до начала клинических исследований с участием детей.
84. Достаточность и объем данных о безопасности исследуемого лекарственного препарата, полученных в клинических исследованиях с участием взрослых определяются в индивидуальном порядке. До начала применения лекарственного препарата у детей достаточные данные об опыте применения этого лекарственного препарата у взрослых могут отсутствовать (например, при исключительно детских показаниях).
85. До начала проведения исследований с участием детей необходимо располагать результатами:
а) исследований токсичности при повторном (многократном) введении лекарственного препарата взрослым животным в течение определенного периода;
б) основного набора (батареи) исследований фармакологической безопасности;
в) стандартного набора исследований генотоксичности.
86. Может потребоваться проведение исследования репродуктивной токсичности, соответствующего возрасту и полу исследуемых детей, для получения сведений о прямых токсических или онтогенетических рисках (например, исследования фертильности и пренатального (постнатального) развития).
87. Проведение клинических исследований эмбриофетального развития необязательно для обоснования проведения таких исследований с участием мужчин и препубертатных женщин.
88. Токсикологические исследования на неполовозрелых животных проводят, если имеющиеся данные о безопасности исследуемого лекарственного препарата, полученные в результате исследований на животных и с участием человека, включая данные об эффектах других лекарственных препаратов этого фармакологического класса, считаются недостаточными для обоснования проведения исследований с участием детей. Если требуется проведение токсикологического исследования, достаточно использование 1 вида неполовозрелых животных, в основном грызунов. При достаточном научном обосновании разрешается проведение доклинических исследований на негрызунах. При краткосрочных фармакокинетических исследованиях с участием детей (например, 1 — 3 дозы лекарственного препарата) проведение предварительных токсикологических исследований на неполовозрелых животных не требуется.
89. В зависимости от показаний к применению, возраста детей и данных о безопасности применения лекарственного препарата, полученных в результате исследований на взрослых животных и с участием человека, до начала краткосрочных клинических исследований эффективности и безопасности с повторным (многократным) введением необходимо рассмотреть вопрос о целесообразности предварительного получения результатов исследований на неполовозрелых животных.
90. Одним из наиболее важных факторов является возраст участников исследования по отношению к продолжительности проведения клинического исследования (то есть период развития, в течение которого участники клинического исследования подвергаются экспозиции лекарственного препарата). Эта оценка позволяет определить необходимость проведения исследований на неполовозрелых животных, установить сроки их проведения по отношению к клиническим исследованиям.
91. При проведении долгосрочных клинических исследований с участием детей, если требуется оценка токсичности в отношении неполовозрелых животных, доклинические исследования необходимо завершить до начала исследований.
92. Проведение долгосрочных исследований токсичности в отношении неполовозрелых животных необходимо в случаях, если детская популяция является основной популяцией, которая получает лекарственный препарат, а в проведенных доклинических исследованиях на животных обнаружены потенциальные онтогенетические риски (токсикологические или фармакологические) для органов-мишеней. Это может быть долгосрочное доклиническое исследование, начатое на животных соответствующего возраста и вида, с соответствующими конечными точками для анализа такого онтогенетического риска (например, 12-месячное исследование на собаках (которое при этом может включать в себя весь период развития собак) или 6-месячное исследование на крысах). У любого из видов животных допускается адаптация данного дизайна доклинического исследования в целях замены им соответствующего стандартного долгосрочного доклинического исследования и отдельного доклинического исследования на неполовозрелых животных.
93. До начала долгосрочных клинических исследований с участием детей необходимо определить целесообразность проведения исследования канцерогенного потенциала. Вместе с тем при отсутствии существенной причины для опасений (например, признаки генотоксичности по результатам разных исследований, наличие проканцерогенного риска, обусловленного механизмом действия действующего вещества, или наличие данных, выявленных при проведении исследований общей токсичности) не требуется проведение исследований канцерогенного потенциала для обоснования проведения клинических исследований с участием детей.
XIV. Исследование иммунотоксичности
лекарственного препарата
94. Новые лекарственные препараты для медицинского применения подлежат оценке в целях подтверждения наличия иммунотоксического потенциала с проведением стандартных токсикологических исследований и дополнительных исследований иммунотоксичности на основе анализа значимости доказательств потенциальной иммунотоксичности лекарственного препарата, включая иммуноопосредованные сигналы, полученные при проведении стандартных токсикологических исследований.
95. Если показано проведение дополнительных исследований иммунотоксичности, их необходимо завершить до экспозиции лекарственного препарата у большой популяции пациентов (например, III фазы клинических исследований).
XV. Исследование фотобезопасности лекарственного препарата
96. Целесообразность и сроки проведения исследования фотобезопасности по отношению к экспозиции лекарственного препарата у человека определяются:
а) фотохимическими свойствами молекулы (например, фотоабсорбция и фотостабильность);
б) сведениями о фототоксическом потенциале химически родственных соединений;
в) распределением в тканях;
г) клиническими или доклиническими проявлениями, свидетельствующими о фототоксичности.
97. Необходимо провести первоначальную оценку фототоксического потенциала исходя из фотохимических свойств и фармакологического (химического) класса. Если оценка всех доступных данных и предлагаемого плана клинических исследований свидетельствует о потенциальном существенном риске фототоксичности для человека, в амбулаторных клинических исследованиях необходимо предусмотреть соответствующие меры защиты.
98. В целях получения сведений о потенциальном фототоксическом риске лекарственного препарата для человека и определения необходимости дальнейшего изучения выполняется доклиническая оценка распределения лекарственного препарата в коже и глазах. При необходимости проводится экспериментальная оценка (доклиническая оценка in vitro или in vivo либо клиническая оценка) фототоксического потенциала до экспозиции лекарственного препарата у большого числа пациентов (III фаза клинических исследований).
99. В качестве альтернативы пошаговому подходу, предусмотренному пунктами 97 и 98 настоящего Руководства, в ходе доклинических или клинических исследований можно провести прямую оценку фототоксического потенциала. Если результаты такого исследования отрицательны, проведение ранней оценки распределения лекарственного препарата в коже и глазах и защитных клинических мер не требуется.
100. Если результаты проведения оценки фототоксичности свидетельствуют о потенциальном фотоканцерогенном риске, у пациентов этот риск обычно в достаточной степени контролируется с применением защитных мер, включающих в себя предостережение в информированном согласии и в информации о зарегистрированном лекарственном препарате. Проведение исследований фотоканцерогенности на грызунах с использованием доступных моделей (например, исследование с участием бесшерстных грызунов) для обоснования разработки лекарственных препаратов считается нецелесообразным и, как правило, не требуется. Если исследования фототоксичности указывают на наличие фотоканцерогенного риска и существует соответствующая методика изучения фотоканцерогенного действия лекарственного препарата, такое исследование необходимо, как правило, завершить до регистрации этого лекарственного препарата, а его результаты необходимо учитывать при оценке риска для человека.
XVI. Доклиническая оценка развития
лекарственной зависимости
101. В отношении действующих на центральную нервную систему лекарственных препаратов независимо от их показания к применению необходимо оценить риск развития лекарственной зависимости. Доклинические исследования должны обосновывать:
а) дизайн клинической оценки потенциала возникновения лекарственной зависимости;
б) определение условий отпуска лекарственного препарата населению;
в) составление информации о лекарственном препарате.
102. При планировании исследований, включающих в себя оценку возникновения лекарственной зависимости разрешается использовать руководства по проведению доклинической оценки риска развития лекарственной зависимости государств-членов.
103. Доклинические данные, полученные на ранних этапах проведения доклинических исследований лекарственного препарата до его первого введения человеку, необходимо учитывать при выявлении ранних индикаторов потенциальной способности лекарственного препарата вызывать развитие лекарственной зависимости. Такие ранние индикаторы включают в себя:
а) фармакокинетический (фармакодинамический) профиль лекарственного препарата, определяющий продолжительность его действия;
б) сходство химической структуры лекарственного препарата с известными лекарственными препаратами, вызывающими лекарственную зависимость;
в) профиль связывания действующего вещества лекарственного препарата с рецепторами;
г) поведенческие (клинические) признаки, установленные при применении лекарственного препарата в доклинических исследованиях in vivo.
104. Если по результатам ранних этапов проведения доклинических исследований потенциал лекарственного препарата в части развития лекарственной зависимости не установлен, расширенные доклинические исследования этого лекарственного препарата на моделях оценки зависимости могут не потребоваться.
105. Если действующее вещество лекарственного препарата проявляет признаки, сходные с известным профилем проявления лекарственной зависимости, или имеет новый механизм действия на центральную нервную систему, для обоснования крупных клинических исследований (например, III фазы), как правило, необходимо провести дополнительные доклинические исследования.
106. Если профиль метаболитов и мишени действия лекарственного препарата у грызунов совпадают с профилем метаболитов и мишенями действия лекарственного препарата у человека, доклиническую оценку риска развития зависимости необходимо провести на грызунах. Разрешается проводить исследования на нечеловекообразных приматах, если выполняются оба следующих условия:
а) есть неоспоримые признаки того, что их участие в доклинических исследованиях поможет спрогнозировать подверженность человека развитию лекарственной зависимости;
б) неприменимы модели изучения на грызунах способности лекарственного препарата вызывать лекарственную зависимость.
107. В целях доклинической оценки риска развития лекарственной зависимости наиболее часто проводятся 3 вида исследований:
а) предпочтение лекарственного препарата;
б) самовведение лекарственного препарата;
в) оценка отмены приема лекарственного препарата.
108. Исследования предпочтения и самовведения лекарственного препарата проводятся как самостоятельные эксперименты. Проведение оценки отмены приема лекарственного препарата разрешается включить в дизайн исследования токсичности с повторным (многократным) введением лекарственного препарата для группы животных с оценкой обратимости восстановления. Максимальная доза, при которой концентрация в плазме в несколько раз превышает концентрацию, полученную при приеме терапевтической клинической дозы у человека, рассматривается как подходящая для такой доклинической оценки риска развития лекарственной зависимости.
XVII. Другие исследования токсичности
лекарственного препарата
109. Если полученные ранее доклинические или клинические данные о лекарственном препарате или родственных лекарственных препаратах указывают на возможные проблемы в отношении безопасности этого лекарственного препарата, целесообразно проведение дополнительных исследований (например, в целях определения потенциальных биомаркеров для изучения механизма действия лекарственного препарата). Проведение дополнительных доклинических исследований (например, для выявления потенциальных биомаркеров изучения механизма действия лекарственного препарата) целесообразно, если предыдущие доклинические или клинические исследования лекарственного препарата или родственных лекарственных препаратов указывали на особые проблемы, связанные с безопасностью лекарственного препарата.
110. Подходы к квалификации примесей и продуктов деградации должны соответствовать правилам по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, утверждаемых Евразийской экономической комиссией. Если для квалификации примеси и продукта деградации необходимо проведение отдельных исследований, их допускается не проводить до начала III фазы клинических исследований. Однако если в рамках разработки и изучения лекарственного препарата происходят изменения, приводящие к образованию нового профиля примесей (например, новый путь синтеза, новый продукт деградации, образовавшийся в результате взаимодействия между компонентами производственной рецептуры), требуется проведение соответствующих исследований квалификации примеси и продукта деградации для обоснования проведения доклинических исследований II фазы или более поздних этапов разработки лекарственного препарата.
XVIII. Исследование токсичности комбинированных
лекарственных препаратов
111. В настоящем разделе описаны исследования токсичности комбинированных лекарственных препаратов, которые упаковываются совместно или вводятся в 1 лекарственной форме (фиксированная комбинация доз). Указанные в настоящем разделе принципы также могут применяться при разработке лекарственных препаратов, которые согласно информации о них будут одновременно применяться с определенным лекарственным препаратом в составе комбинированной терапии (даже если он не будет входить в фиксированную комбинацию доз) и о безопасности комбинирования которых получены минимальные клинические сведения.
112. Положения настоящего раздела применяются при проведении изучения токсичности комбинированных лекарственных препаратов, содержащих в своем составе следующие комбинации действующих веществ:
а) лекарственный препарат представляет собой комбинацию 2 действующих веществ или более, находящихся на позднем этапе разработки (то есть эти действующие вещества являются соединениями со значительным опытом клинического применения, для которых имеются данные по III фазе клинических исследований, и (или) эти комбинации содержатся в зарегистрированных лекарственных препаратах);
б) лекарственный препарат представляет собой комбинацию, в которой 1 действующее вещество или более находятся на позднем этапе разработки и 1 действующее вещество или более находятся на раннем этапе разработки (то есть вторая часть комбинации представляет собой соединения с ограниченным опытом клинического применения, для которых имеются только данные по II фазе клинических исследований или данные по более ранним фазам клинических исследований);
в) лекарственный препарат представляет собой комбинацию, в которой все действующие вещества находятся на раннем этапе разработки.
113. В отношении большинства комбинаций действующих веществ, содержащих 2 действующих вещества, находящихся на позднем этапе разработки и имеющих достаточный опыт совместного клинического применения, в отсутствие существенных опасений относительно токсического действия (например, аналогичная токсичность в отношении органа-мишени) проведение доклинических исследований комбинации для обоснования проведения клинических исследований и регистрации лекарственного препарата не требуется. Опасения относительно токсического действия устанавливаются исходя из границ безопасности лекарственного препарата и возможности мониторинга нежелательных реакций у человека. Если исследование проводится в целях поиска причины возникших при разработке комбинированного лекарственного препарата опасений относительно его токсического действия, такое исследование, как правило, необходимо завершить до начала клинических исследований комбинированного лекарственного препарата.
114. Проведение доклинических исследований для обоснования проведения небольших, относительно краткосрочных клинических исследований (например, исследований II фазы исследования продолжительностью до 3 месяцев) не требуется в случае, если в отношении лекарственных препаратов, содержащих 2 действующих вещества и более, находящихся на позднем этапе разработки, не имеется достаточного опыта совместного клинического применения, но на основании имеющихся данных нет причин для существенных опасений относительно токсического действия. Вместе с тем для проведения продолжительных или крупномасштабных клинических исследований, а также для регистрации лекарственного препарата проведение доклинических исследований указанных комбинаций обязательно.
115. Проведение токсикологических исследований комбинированных лекарственных препаратов для обоснования проведения их клинических исследований продолжительностью до 1 месяца с проверкой безопасности таких комбинированных лекарственных препаратов не требуется в случае, если лекарственный препарат является комбинацией веществ, находящихся на раннем этапе разработки и имеющих опыт клинического применения, с веществами, находящимися на позднем этапе разработки и не имеющими установленного токсического воздействия. При этом клиническое исследование таких комбинаций не должно по продолжительности превышать клинический опыт применения отдельных компонентов. Если клинические исследования проводятся на более позднем этапе разработки комбинированного лекарственного препарата или имеют большую продолжительность, их необходимо обосновать доклиническими токсикологическими исследованиями комбинации.
116. В отношении лекарственных препаратов, содержащих комбинацию 2 действующих веществ и более, находящихся на раннем этапе разработки, для обоснования проведения клинических исследований необходимо провести доклинические токсикологические исследования такой комбинации.
117. Если в отношении отдельных компонентов лекарственного препарата из комбинации 2 действующих веществ и более проведена полная программа доклинической разработки, для обоснования проведения клинических исследований такого комбинированного лекарственного препарата требуется проведение доклинического токсикологического исследования продолжительностью эквивалентной продолжительности клинического исследования, но не более 90 дней введения лекарственного препарата.
Для одобрения регистрации комбинированного лекарственного препарата необходимо представление результатов указанного 90-дневного токсикологического исследования. Однако в случае кратковременной продолжительности планируемого клинического применения лекарственного препарата в целях его регистрации разрешается представление результатов проведения токсикологического исследования комбинации действующих веществ меньшей продолжительности.
117. Если в отношении отдельных компонентов лекарственного препарата из комбинации 2 действующих веществ и более, находящихся на раннем этапе разработки, проведена полная программа доклинической разработки, для обоснования проведения клинических исследований такого комбинированного лекарственного препарата требуется проведение доклинического токсикологического исследования продолжительностью эквивалентной продолжительности клинического исследования, но не более 90 дней введения лекарственного препарата.
Для одобрения регистрации комбинированного лекарственного препарата необходимо представление результатов указанного 90-дневного токсикологического исследования. Однако в случае кратковременной продолжительности планируемого клинического применения лекарственного препарата в целях его регистрации разрешается представление результатов проведения токсикологического исследования комбинации действующих веществ меньшей продолжительности.
118. Дизайн доклинических исследований, рекомендуемых для определения характеристик комбинации действующих веществ, зависит от фармакологического, токсикологического и фармакокинетического профилей отдельных компонентов, показаний к применению, целевой популяции пациентов и имеющихся клинических данных.
119. Доклинические исследования комбинации действующих веществ в составе лекарственного препарата необходимо, как правило, проводить с участием 1 релевантного вида животных. При выявлении непредвиденной токсичности разрешается проведение дополнительных исследований.
120. Если в отношении отдельных компонентов комбинированного лекарственного препарата полная программа доклинической разработки не выполнена, разрешается провести полную программу доклинических токсикологических исследований исключительно в отношении всей комбинации действующих веществ в составе лекарственного препарата при условии, что отдельные компоненты этой комбинации предназначены исключительно для применения в составе комбинированного лекарственного препарата.
121. Если отдельные компоненты комбинации действующих веществ были изучены в соответствии с актами органов Союза в сфере обращения лекарственных средств, для обоснования проведения клинических исследований и регистрации лекарственного препарата проведение исследований генотоксичности, фармакологической безопасности и канцерогенности комбинации, как правило, не требуется.
122. Если популяция пациентов включает в себя женщин с детородным потенциалом, а результаты исследований отдельных компонентов свидетельствуют об эмбриофетальном риске, проводить исследования комбинации не требуется, поскольку уже выявлена потенциальная опасность для эмбриофетального развития человека.
123. Если результаты проведения доклинических исследований эмбриофетального развития свидетельствуют о том, что ни один из компонентов комбинации действующих веществ не несет онтогенетического риска для человека, при отсутствии опасений (основанных на свойствах отдельных компонентов), что их комбинация может представлять опасность для человека, проведение исследований комбинации действующих веществ не требуется.
124. Если отдельные компоненты комбинации действующих веществ изучены при проведении доклинических исследований эмбриофетального развития, но требуется проведение эмбриофетальных исследований комбинации действующих веществ, результаты таких исследований необходимо представить для обоснования регистрации лекарственного препарата.
Приложение N 1
к Руководству по доклиническим
исследованиям безопасности
в целях проведения клинических
исследований и регистрации
лекарственных препаратов
ТРЕБОВАНИЯ
К СОСТАВЛЕНИЮ ДОКУМЕНТОВ О ДОКЛИНИЧЕСКИХ ИССЛЕДОВАНИЯХ
ДЛЯ СМЕШАННЫХ РЕГИСТРАЦИОННЫХ ДОСЬЕ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
I. Область применения
1. Ряд лекарственных препаратов, применяемых в медицинской практике в течение длительного времени, содержит действующие вещества, данные о доклиническом исследовании которых ограничены или отсутствуют. Для оценки рисков, связанных с применением таких лекарственных препаратов, и во избежание проведения повторных экспериментов на животных в настоящем документе определяется минимальный объем доклинических исследований таких лекарственных препаратов.
2. Положения настоящего документа распространяются на лекарственные препараты, действующее вещество которых, относится к группе химических действующих веществ с известным химическим строением.
3. Положения настоящего документа не распространяются на биологические, биотехнологические и лекарственные растительные препараты.
4. Под смешанным регистрационным досье лекарственного препарата понимается вид регистрационного досье лекарственного препарата, в котором модули 4 и 5 представляют собой комбинацию результатов собственных доклинических и (или) клинических исследований и обзора фармако-токсикологических свойств лекарственного препарата на основе опубликованных в научной медицинской литературе сведений с указанием библиографических ссылок (включая научные работы, клинические исследования, данные пострегистрационного опыта, полученного по результатам широкого клинического применения лекарственного препарата человеком). Данные, содержащиеся в смешанном регистрационном досье лекарственного препарата позволяют оценить безопасность применения такого лекарственного препарата.
II. Документы о доклинических исследованиях
1. Общие положения
5. Проведение доклинических исследований, как правило, не требуется в случае, если имеется достаточный надлежащим образом документированный опыт клинического применения для установления всех аспектов клинической эффективности и безопасности лекарственного препарата.
6. Доклинические исследования необходимо проводить в случае, если фармакологический класс веществ, к которому относится лекарственный препарат, или опыт клинического применения лекарственного препарата вызывают обоснованные опасения о небезопасности применения такого лекарственного препарата или в случае, если возникает подозрение в отношении небезопасности его применения.
7. Отсутствие отдельных доклинических исследований (в частности, исследований репродуктивной токсичности, генотоксичности и канцерогенности) является основанием для наличия у уполномоченного органа (экспертной организации) государства — члена Евразийского экономического союза опасений о небезопасности лекарственного препарата. Если лекарственный препарат способен вызывать токсические эффекты, которые трудно или невозможно обнаружить в рамках клинического изучения этого лекарственного препарата, требуется проведение доклинических исследований.
2. Отдельные виды доклинических исследований
Токсичность при однократном и повторном (многократном)
введении лекарственного препарата
8. Допускается не проводить исследование токсичности при однократном и повторном (многократном) введении лекарственного препарата, а также исследование его фармакологических свойств (включая исследование фармакологической безопасности и фармакокинетики) в случае, если планируется проведение регистрации данного лекарственного препарата с представлением смешанного регистрационного досье.
Репродуктивная и онтогенетическая токсичность
9. Проведение доклинических исследований влияния лекарственного препарата на фертильность и общую репродуктивную функцию не требуется при отсутствии опасений о небезопасности лекарственного препарата.
10. Необходимо оценить потенциал влияния репродуктивной токсичности лекарственного препарата на эмбриофетальное и перинатальное (постнатальное) развитие человека. Необходимо принимать во внимание, что данные о репродуктивной токсичности большинства действующих веществ могут быть доступны в виде фармакотоксикологических сведений и могут содержаться в научных публикациях, но при этом таких данных недостаточно для надлежащей оценки безопасности лекарственного препарата.
11. Проведение исследований эмбриофетальной токсичности лекарственного препарата и его влияния на перинатальное (постнатальное) развитие не требуется для каждого следующего случая:
а) получены достаточные данные об экспозиции лекарственного препарата у беременных и новорожденных;
б) лекарственный препарат не предназначен для применения женщинами с детородным потенциалом;
в) лекарственный препарат не предназначен для применения во время беременности и в период грудного вскармливания.
Генотоксичность
12. Необходимо оценить генотоксический потенциал действующего вещества лекарственного препарата.
13. Необходимо принимать во внимание, что данные о генотоксичности большинства действующих веществ могут быть доступны в виде фармакотоксикологических сведений и могут содержаться в научных публикациях, но при этом таких данных недостаточно для надлежащей оценки безопасности лекарственного препарата. В случае если невозможно провести полноценную оценку мутагенности и (или) хромосомных повреждений у человека при применении данного лекарственного препарата, требуется проведение дополнительных исследований на генотоксичность.
14. В ряде случаев разрешается экстраполировать генотоксические свойства других веществ (например, цитостатических средств), принадлежащих к определенному классу анатомо-терапевтическо-химической классификации, на действующее вещество лекарственного препарата, принадлежащего к этому же фармакологическому классу. В таких случаях проведение исследований генотоксичности лекарственного препарата не требуется.
Канцерогенность
15. Проведение экспериментальных исследований канцерогенности не требуется в случае, если отсутствуют подозрения в отношении наличия у действующего вещества канцерогенного потенциала.
16. В случае возникновения подозрения в отношении наличия у действующего вещества канцерогенного потенциала при принятии разработчиком лекарственного препарата решения о необходимости проведения собственных экспериментальных исследований канцерогенности необходимо учитывать следующие обстоятельства:
а) влияние положительного результата таких исследований на оценку соотношения «польза — риск»;
б) прогнозируемость индукции опухолей на основании результатов предыдущих исследований действующих веществ с аналогичной молекулярной структурой и (или) механизмом действия;
в) обоснованность подозрения в отношении канцерогенного действия лекарственного препарата (наличие положительных результатов исследований его генотоксичности) и возможность устранения этого подозрения посредством проведения дополнительных исследований его генотоксичности (главным образом in vivo);
г) обоснованность подозрения в отношении канцерогенного действия лекарственного препарата, подтвержденного положительными результатами эпидемиологических наблюдений у человека (например, эстроген-индуцированные опухоли молочной железы);
д) наличие в регистрационном досье лекарственного препарата научных данных необходимых для устранения подозрения в отношении канцерогенного потенциала действующего вещества лекарственного препарата.
Токсикокинетические данные требуются для представления в составе регистрационного досье лекарственного препарата только при проведении новых исследований канцерогенности на животных.
III. Доклинический обзор
17. Специалист производителя лекарственных средств, ответственный за подготовку модуля 2 регистрационного досье лекарственного препарата, обязан проанализировать совокупность доступных сведений, подтверждающих допустимый уровень безопасности действующего вещества. В случае наличия действующих веществ с недостающими токсикологическими данными этот специалист обязан проанализировать результаты соответствующих фармакологических и токсикологических исследований лекарственного препарата, указав ссылки на научную литературу, и (или) представить в регистрационном досье лекарственного препарата научное обоснование, подтверждающее допустимый уровень безопасности действующего вещества (с учетом сведений о широком клиническом применении этого действующего вещества). При выполнении такого анализа и интерпретации результатов исследований специалист должен учитывать степень значимости возможных отклонений ранее выполненных исследований от требований к выполнению доклинических исследований (предусмотренных Правилами надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81).
18. В разделы 4.6 и 5.3 общей характеристики лекарственного препарата, представляемой в составе модуля 1 регистрационного досье лекарственного препарата, необходимо включить формулировки об отсутствии собственных экспериментальных данных о безопасности таких лекарственных препаратов.
Приложение N 2
к Руководству по доклиническим
исследованиям безопасности
в целях проведения клинических
исследований и регистрации
лекарственных препаратов
ТРЕБОВАНИЯ
К МИНИМАЛЬНОЙ ПРОДОЛЖИТЕЛЬНОСТИ ДОКЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ
ПРИ ПОВТОРНОМ (МНОГОКРАТНОМ) ВВЕДЕНИИ ЛЕКАРСТВЕННОГО
ПРЕПАРАТА, НЕОБХОДИМЫХ ДЛЯ ОБОСНОВАНИЯ ПРОВЕДЕНИЯ
КЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ РАЗЛИЧНОЙ ПРОДОЛЖИТЕЛЬНОСТИ
|
Максимальная продолжительность клинического исследования |
Минимальная продолжительность исследований токсичности при повторном (многократном) введении лекарственного препарата для обоснования продолжительности проведения клинических исследований |
|
|
грызуны |
негрызуны |
|
| До 2 недель |
2 недели <1> |
2 недели <1> |
|
От 2 недель До 6 месяцев |
такая же, как в клинических исследованиях <2> |
такая же, как в клинических исследованиях <2> |
| Свыше 6 месяцев |
6 месяцев <2>, <3> |
9 месяцев <2>, <3>, <4> |
<1> Возможной альтернативой 2-недельным исследованиям для обоснования проведения клинических исследований при однократном введении лекарственного препарата является расширенное исследование токсичности при однократном введении лекарственного препарата. В рамках расширенного исследования токсичности при однократном введении лекарственного препарата необходимо, как правило, оценить данные по гематологии, биохимии, некропсии и гистопатологии (только для контроля и максимальной дозы, если при максимальной дозе не обнаруживается патология, обусловленная применением препарата) после однократного введения лекарственного препарата с последующим изучением спустя 2 недели для оценки отсроченной токсичности и (или) восстановления. Стандартный дизайн исследования на грызунах предусматривает использование 10 особей каждого пола на группу, оцениваемых на следующий день после введения, и 5 особей каждого пола для дозы (доз), оцениваемой на 14-й день после введения. Стандартный дизайн исследований на негрызунах предусматривает использование 3 особей каждого пола во всех группах для оценки на 2-й день и 2 особей каждого пола при дозе (дозах), оцениваемых на 14-й день. Клинические исследования продолжительностью менее 14 дней разрешается обосновывать исследованиями токсичности с той же продолжительностью.
<2> Клинические исследования продолжительностью более 3 месяцев разрешается начинать при наличии результатов 3-месячного исследования на грызунах и 3-месячного исследования на негрызунах при выполнении следующих условий:
уполномоченному органу (экспертной организации) государства — члена Евразийского экономического союза представлено обоснование такой программы доклинической и клинической разработки;
представлены полные результаты исследований токсичности при повторном (многократном) введении грызунам и негрызунам прежде, чем продолжительность дозирования лекарственного препарата в клиническом исследовании превысит 3 месяца.
При серьезных или угрожающих жизни показаниях или в индивидуальном порядке такое продление возможно при наличии результатов полностью завершенных исследований токсичности при повторном (многократном) введении грызунам и результатов прижизненных исследований и данных некропсии в исследовании на негрызунах. Полные патогистологические данные исследований на негрызунах необходимо получить в течение последующих 3 месяцев.
<3> В некоторых случаях основной популяцией являются дети, а имеющиеся доклинические исследования на животных (токсикологические или фармакологические) указывают на потенциальные онтогенетические опасения в отношении органов-мишеней. В этих случаях при определенных обстоятельствах могут потребоваться долгосрочные исследования токсичности, начатые на неполовозрелых животных.
<4> Исследования 6-месячной продолжительности на негрызунах считаются достаточными. Вместе с тем в случае, если проведены исследования большей продолжительности, проведение дополнительных 6-месячных исследований не требуется. Исследования на негрызунах 6-месячной продолжительности также достаточны в следующих случаях:
иммуногенность или непереносимость лекарственного препарата может исказить результаты более длительных исследований;
пациенты подвергаются многократной краткосрочной экспозиции даже в условиях клинического исследования продолжительностью свыше 6 месяцев (например, при нерегулярном лечении мигрени, эректильной дисфункции или простого герпеса);
лекарственный препарат предназначен для длительного применения для снижения риска рецидива онкологических заболеваний;
лекарственный препарат предназначен для применения по показаниям пациентами с короткой ожидаемой продолжительностью жизни.
Приложение N 3
к Руководству по доклиническим
исследованиям безопасности
в целях проведения клинических
исследований и регистрации
лекарственных препаратов
ТРЕБОВАНИЯ
К МИНИМАЛЬНОЙ ПРОДОЛЖИТЕЛЬНОСТИ ДОКЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ
ПРИ ПОВТОРНОМ (МНОГОКРАТНОМ) ВВЕДЕНИИ ЛЕКАРСТВЕННОГО
ПРЕПАРАТА, НЕОБХОДИМЫХ ДЛЯ ОБОСНОВАНИЯ ПРОДОЛЖИТЕЛЬНОСТИ
ЕГО ПРИМЕНЕНИЯ ЧЕЛОВЕКОМ (ПРИ РЕГИСТРАЦИИ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА)
|
Продолжительность применения по показанию |
Грызуны |
Негрызуны |
| До 2 недель |
1 месяц |
1 месяц |
| Свыше 2 недель до 1 месяца |
3 месяца |
3 месяца |
| Свыше 1 месяца до 3 месяцев |
6 месяцев |
6 месяцев |
| Свыше 3 месяцев |
6 месяцев <1> |
9 месяцев <1>, <2> |
<1> В некоторых случаях основной популяцией являются дети, а имеющиеся доклинические исследования на животных (токсикологические или фармакологические) указывают на потенциальные онтогенетические опасения в отношении органов-мишеней. В этих случаях при определенных обстоятельствах могут потребоваться долгосрочные исследования токсичности, начатые на неполовозрелых животных.
<2> Исследования 6-месячной продолжительности на негрызунах считаются достаточными. Вместе с тем в случае, если проведены исследования большей продолжительности, проведение дополнительных 6-месячных исследований не требуется. Исследования на негрызунах 6-месячной продолжительности также достаточны в следующих случаях:
иммуногенность или непереносимость лекарственного препарата может исказить результаты более длительных исследований;
пациенты подвергаются многократной краткосрочной экспозиции даже в условиях клинического исследования продолжительностью свыше 6 месяцев (например, при нерегулярном лечении мигрени, эректильной дисфункции или простого герпеса);
лекарственный препарат предназначен для длительного применения для снижения риска рецидива онкологических заболеваний;
лекарственный препарат предназначен для применения по показаниям пациентами с короткой ожидаемой продолжительностью жизни.
Приложение N 4
к Руководству по доклиническим
исследованиям безопасности
в целях проведения клинических
исследований и регистрации
лекарственных препаратов
ТРЕБОВАНИЯ
К ПРОВЕДЕНИЮ ДОКЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ, НЕОБХОДИМЫХ
ДЛЯ ОБОСНОВАНИЯ ОБЪЕМА ПРОВЕДЕНИЯ ПОИСКОВЫХ
КЛИНИЧЕСКИХ ИССЛЕДОВАНИЙ
|
Поисковые клинические исследования |
Доклинические исследования |
|||
|
вводимая доза |
начальная и максимальная дозы |
исследование фармакологических свойств |
исследования общетоксических свойств <1> |
исследование генотоксичности <2> (прочие исследования) |
|
Подход 1-й. Суммарная доза удовлетворяет всем следующим критериям: фармакологически активной дозы (выраженной в мг/кг для внутривенного введения и в мг/м2 для перорального введения) |
максимальная и начальная дозы могут быть одинаковыми, но не должны превышать суммарную кумулятивную дозу в 100 мкг | необходимо провести профилирование связывания с мишенями (рецепторами) in vitro. Для обоснования выбора дозы для человека необходимо располагать результатами установления соответствующих характеристик первичной фармакологии (механизма действия и (или) эффектов) на фармакологически значимой модели | расширенное исследование токсичности при однократном введении <3>, <4> на одном виде животных (обычно грызунах) при планируемом пути введения с данными по токсикокинетике или при внутривенном введении. Разрешается использовать максимальную дозу, в 1 000 раз превышающую клиническую дозу, выраженную в мг/кг для внутривенного введения и в мг/м2 поверхности тела для перорального введения | проведение исследований генотоксичности не требуется, однако в досье клинического исследования необходимо включить все проведенные исследования и оценку структурно-функциональной зависимости. Необходимо представить соответствующие фармакокинетические данные и дозиметрические показатели высокорадиоактивных веществ (например, веществ для ПЭТ-визуализации) |
|
Подход 2-й. Выполнение всех следующих критериев: суммарная кумулятивная доза каждая доза каждая доза фармакологически активной дозы |
максимальная суточная и начальная дозы могут быть одинаковыми, но не должны превышать суммарную кумулятивную дозу в 100 мкг | необходимо провести профилирование связывания с мишенями (рецепторами) in vitro. Для обоснования выбора дозы для человека необходимо располагать результатами установления соответствующих характеристик первичной фармакологии (механизма действия и (или) эффектов) на фармакологически значимой модели | 7-дневное исследование токсичности при повторном (многократном) введении на одном виде животных (обычно грызунах) при планируемом пути введения с данными по токсикокинетике или при внутривенном введении. Необходимо включить данные о гематологии, биохимических исследованиях материала, некропсии и гистопатологии. Разрешается использовать максимальную дозу, в 1 000 раз превышающую клиническую дозу, выраженную в мг/кг для внутривенного введения и в мг/м2 для перорального введения | проведение исследований генотоксичности не требуется, однако в досье клинического исследования необходимо включить все проведенные исследования и оценку структурно-функциональной зависимости. Необходимо представить соответствующие фармакокинетические данные и дозиметрические показатели высокорадиоактивных веществ (например, веществ для ПЭТ-визуализации) |
|
Подход 3-й. Исследования с однократным введением субтерапевтических доз или доз предполагаемого терапевтического диапазона |
начальную дозу необходимо определить исходя из характера токсических данных, выявленных у наиболее чувствительного вида животных, и предполагаемой фармакологически активной дозы. Необходимо учесть вопросы выбора начальной дозы для человека исходя из требований соответствующих руководств, применяемых в государствах — членах Евразийского экономического союза (далее — государства-члены). Максимальной дозой может быть доза, обеспечивающая достижение |
необходимо провести профилирование связывания с мишенями (рецепторами) in vitro. Для обоснования выбора дозы у человека необходимо располагать результатами установления соответствующих характеристик первичной фармакологии (механизма действия и (или) эффектов) на фармакологически значимой модели. Основной набор (батарея) исследований фармакологической безопасности | расширенное исследование токсичности при однократном введении грызунам и негрызунам <3> при клиническом пути введения с данными по токсикокинетике, гематологии, клинической биохимии, некропсии и гистопатологии. В этом случае максимальной дозой должна быть максимальная переносимая доза, максимальная достижимая доза или предельная доза | тест Эймса (или альтернативный метод, если проведение теста Эймса невозможно (например, в отношении антибактериального лекарственного препарата)) |
|
Подход 4-й. Дозирование до 14 дней в терапевтическом диапазоне, но не с целью оценки клинической максимальной переносимой дозы |
При выявлении токсичности у 2 видов животных необходимо учесть вопросы выбора начальной дозы для человека исходя из требований соответствующих руководств, применяемых в государствах-членах. Если ни у одного вида животных токсичность не выявлена (то есть NOAEL являлись максимальными исследованными дозами, которые не были ограничены другим образом (например, не являлись максимальной достижимой дозой)) или выявлена только у одного вида животных, клинической начальной дозой должна стать доза, обеспечивающая прогнозируемое клиническое значение AUC (основанное или на межвидовом фармакокинетическом моделировании, или на преобразовании по мг/м2), что примерно равно 1/50 AUC при NOAEL у вида животных с меньшей экспозицией. Другие вопросы выбора начальной дозы для человека (например, исходя из прогнозируемой фармакодинамической активности) необходимо учитывать исходя из требований соответствующих руководств, применяемых в государствах-членах. В отсутствие токсичности у 2 видов животных максимальная клиническая доза не должна превышать 1/10 меньшей экспозиции (AUC), достигнутой у любого из видов животных при максимальной испытанной дозе. Если токсичность проявляется только у одного вида животных, максимальная клиническая доза не должна превышать NOAEL у того вида животных, у которого отмечалась токсичность, или |
необходимо провести профилирование связывания с мишенями (рецепторами) in vitro. Для обоснования выбора дозы для человека необходимо располагать результатами установления соответствующих характеристик первичной фармакологии (механизма действия и (или) эффектов) на фармакологически значимой модели. Основной набор (батарея) исследований фармакологической безопасности в дозах, совпадающих с дозами, использованными в токсикологических исследованиях | 2-недельные исследования токсичности с повторным (многократным) введением грызунам и негрызунам с оценкой стандартных параметров и выбором дозы для животных на основе кратности экспозиции от ожидаемой клинической AUC при максимальной дозе | тест Эймса (или альтернативный метод, если проведение теста Эймса невозможно (например, в отношении антибактериального лекарственного препарата)) и тест (in vitro или in vivo), способный обнаружить повреждение хромосом у млекопитающих |
|
Подход 5-й: дозирование до 14 дней, не превышающее продолжительность дозирования у негрызунов в терапевтическом диапазоне, но не с целью оценки клинической максимальной переносимой дозы |
прогнозируемая величина экспозиции при начальной дозе не должна превышать 1/50 NOAEL у более чувствительного вида животных в мг/м2. Вопросы выбора начальной дозы для человека необходимо учитывать исходя из требований соответствующих руководств, применяемых в государствах-членах. Максимальная экспозиция у человека не должна превышать AUC при NOAEL у негрызунов или |
необходимо провести профилирование связывания с мишенями (рецепторами) in vitro. Для обоснования выбора дозы для человека необходимо располагать результатами установления соответствующих характеристик первичной фармакологии (механизма действия и (или) эффектов) на фармакологически значимой модели. Основной набор (батарея) исследований фармакологической безопасности в дозах, совпадающих с дозами, использованными в токсикологических исследованиях | стандартное 2-недельное исследование токсичности при повторном (многократном) введении грызунам (при обосновании того, что грызуны являются релевантным видом). Максимальная доза должна быть максимальной переносимой дозой, максимальной достижимой дозой или предельной дозой. Подтверждающее исследование на негрызунах (n = 3) в дозе, равной ожидаемой NOAEL-экспозиции у грызунов, продолжительностью не менее 3 дней и по меньшей мере с планируемой продолжительностью клинического исследования или исследование с эскалацией дозы у негрызунов продолжительностью не менее 3 дней и по меньшей мере планируемой продолжительностью клинического исследования при ожидаемой NOAEL-экспозиции у грызунов | тест Эймса (или альтернативный метод, если проведение теста Эймса невозможно (например, в отношении антибактериального лекарственного препарата)) и тест (in vitro или in vivo), способный обнаруживать повреждение хромосом у млекопитающих. В случае оценки in vivo ее разрешается провести в рамках токсикологического исследования на грызунах |
<1> Исследования общетоксических свойств необходимо проводить в соответствии с требованиями Правил надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81.
<2> Дизайн и выбор дозы для исследований генотоксичности.
<3> В рамках расширенного исследования токсичности при однократном введении лекарственного препарата необходимо, как правило, оценить данные по гематологии, биохимии, некропсии и гистопатологии (только для контроля и максимальной дозы, если при максимальной дозе не обнаруживается патология, обусловленная применением препарата) лекарственного препарата после однократного введения с последующим изучением спустя 2 недели для оценки отсроченной токсичности и (или) восстановления. Стандартный дизайн исследований на грызунах предусматривает использование 10 особей каждого пола на группу, оцениваемых на следующий день после введения, и 5 особей каждого пола для дозы (доз), оцениваемой на 14-й день после введения лекарственного препарата. Стандартный дизайн исследований на негрызунах предусматривает использование 3 особей каждого пола во всех группах для оценки на 2 день и 2 особей каждого пола при дозе (дозах), оцениваемых на 14-й день.
<4> При подходе с исследованием микродоз разрешается проведение оценки обратимости (отсроченной токсичности) на 14-й день после однократного введения. Используемая доза необязательно должна быть максимальной, но она должна превышать клиническую дозу в 100 раз.
<5> При отсутствии нежелательных реакций в клинических исследованиях разрешается эскалация дозы выше AUC, если реакции, выявленные в токсикологических исследованиях, поддаются мониторингу, являются обратимыми и имеют низкую степень тяжести у человека.
Под термином «Надлежащая клиническая практика» (Good Сlinical Рractice, GCP) понимают стандарт клинических исследований, охватывающий планирование, проведение, завершение, проверку, анализ результатов, составление отчетов и ведение документации, который обеспечивает научную значимость исследований, их этическую приемлемость и полную документированность клинических характеристик изучаемого лекарственного препарата.
Как неоднократно отмечалось в отечественной и зарубежной литературе, а также в документах ВОЗ, потребительские свойства лекарственных препаратов – эффективность, безопасность и фармацевтические аспекты качества – обеспечиваются благодаря соблюдению важнейших отраслевых правил, иначе говоря, стандартов или кодексов GLP, GCP и GMP в процессе их разработки, испытания и производства.
В последние годы журнал «Фарматека» уделял особое внимание проблемам внедрения правил GMP в отечественной индустрии лекарств, и этот стандарт рассматривался достаточно подробно*. Недавно был опубликован обзор международных правил GLP в сравнении с российскими правилами доклинических исследований фармакологических веществ**. Но обзорный материал, посвященный правилам GCP, в журнале опубликован не был, хотя организационные и этические аспекты проведения клинических испытаний новых препаратов затрагивались во многих статьях [1–6]. Обзор по правилам GCP позволил бы обобщить и подвести итоги предыдущих публикаций по организации клинических испытаний. Кроме того, он мог бы привлечь внимание к существованию прямой связи между правилами GCP и GMP. Речь идет о дополнениях к правилам GMP для изготовления экспериментальных препаратов (см. ниже).
Международные руководства по клинической практике
В настоящее время на международном уровне действуют 4 основных руководящих документа, определяющие порядок планирования и проведения клинических испытаний лекарственных препаратов. Хельсинская декларация Всемирной медицинской ассоциации [4, 7] определяет основополагающие этические принципы проведения исследований в данной области. Последние расширены и уточнены в рекомендациях Совета международных организаций медицинских наук (CIOMS)*. Наряду с этим имеются два широко признанных в мире кодекса GCP, устанавливающие организационно-методические требования к исследованиям. Первый был выпущен Всемирной организацией здравоохранения (ВОЗ) в 1995 г. [8], второй, разработанный в рамках ICH (кодовое обозначение E6, т.е. шестой документ в разделе «Эффективность» – Efficacy), вступил в силу в 1997 г. [9]. Оба руководства содержат ссылки на Хельсинскую декларацию, ее полный текст дан в виде приложения к документу ВОЗ.
Хельсинская декларация об этических принципах медицинских исследований, проводимых на людях, впервые была опубликована в 1964 г. С того времени она неоднократно уточнялась, однако ее основные положения остались неизменными. Декларация содержит руководящие принципы и рекомендации, адресованные каждому врачу, в отношении этики и прав человека, а также научных и медицинских норм в клинических исследованиях. В соответствии с ней миссия врача заключается в сохранении здоровья людей, а целью биомедицинских исследований на человеке является улучшение диагностических, терапевтических и профилактических методов.
С конца 70-х гг. прошлого века CIOMS разрабатывал международные методические документы, направленные на внедрение принципов Хельсинской декларации в различных странах с учетом местных особенностей культуры, социально-экономических условий, традиций, законов и правил. Эта работа была завершена выходом нескольких руководств, в частности, по этическому обзору эпидемиологических исследований (1991) и этическим принципам биомедицинских исследований на людях (1993) [10–12]. Данные базовые международные руководства, получившие широкое признание, – результат длительных консультаций с участием широкого круга специалистов из индустриальных и развивающихся стран, представлявших министерства здравоохранения, медицинские исследовательские учреждения, этику, философию и право.
ВОЗ активно содействовала всеобщему признанию принципов Хельсинской декларации. Ее основные положения были отражены в документе ВОЗ «Принципы клинической оценки лекарств« уже в 1968 г. [13]. В 1975 г. были опубликованы более подробные рекомендации ВОЗ по оценке лекарств [14], которыми эти положения закреплялись. Рекомендации содержали прямые ссылки на Хельсинскую декларацию. В дальнейшем эти документы легли в основу руководства ВОЗ по GCP.
Правила GСP, также как и правила GLP и GMP, впервые появились в США. Первый вариант правил GСP был опубликован Администрацией по пищевым и лекарственным продуктам (FDA) в 1978 г. в форме проекта. С 1980 г. начались инспекционные проверки хода клинических испытаний на соответствие этому стандарту – вначале в США, а затем и за пределами страны. В 1987 г правила GСP были приняты во Франции, в 1989 г. – в cкандинавских странах и Японии. В 1990 г. Комиссией ЕC были опубликованы так называемые Европейские правила GСP, получившие официальный статус во всех странах Сообщества в 1991 г.
Следование стандарту GСP при проведении ключевых или определяющих (pivotal) клинических испытаний является одним из важнейших условий регистрации новых лекарственных препаратов в индустриальных странах. Результаты исследований, выполненных без соблюдения правил GСP, могут быть приняты к рассмотрению лишь в качестве дополнительных (supporting data). Необходимо подчеркнуть, что исследования, изначально спланированные и проведенные без соблюдения правил GСP, не могут быть приведены к этому стандарту ретроспективно, например, путем какой-либо обработки данных.
Правила GCP ВОЗ и ICH
Правила надлежащей клинической практики ВОЗ были подготовлены в 1991–93 гг. С этой целью было проведено несколько совещаний, в которых участвовали сотрудники органов нормативного контроля лекарств, представители фармацевтической промышленности и ученые-медики из Бельгии, Бразилии, Дании, Замбии, Индонезии, Китая, России*, США, Швеции и Японии. Выработанный в ходе совещаний проект Правил рассылался на согласование органам государственного управления, научным учреждениям и производителям лекарств во многих странах мира. Окончательный вариант документа был утвержден в 1993 г. на 6 заседании Комитета экспертов ВОЗ по использованию основных лекарств и опубликован в качестве приложения к докладу Комитета в 1995 г. Как и другие официально опубликованные материалы экспертных комитетов, доклад с приложениями прошел процедуру рассмотрения на заседании Исполкома и был одобрен этим руководящим органом ВОЗ. Таким образом, документ ВОЗ отражает глобальный консенсус министерств здравоохранения всех 190 с лишним стран-членов Организации.
Международная конференция по гармонизации требований к лекарствам (ICН)* разрабатывала свои рекомендации по GCP в период с 1992 по 1996 гг. К их подготовке привлекались многие эксперты, участвовавшие в разработке документа ВОЗ. В итоге, оба документа согласуются друг с другом в части принципов, хотя между ними имеются определенные различия. Правила ICН являются результатом процесса гармонизации, в котором участвуют контрольно-разрешительные органы и инновационная фармацевтическая промышленность Евросоюза, США и Японии (отсюда одно из названий конференции – «трехсторонняя инициатива»).
Иначе говоря, все методические материалы ICН, в т.ч. и правила GСP, отражают и гармонизируют существующие на текущий момент нормативные документы и сложившуюся практику 17 ведущих индустриальных государств, включающих США, страны Западной Европы и Японию. По сути, они предназначены для использования в странах так называемого «золотого миллиарда», входящих в первую двадцатку индустриальных держав**. По своему географическому охвату трехсторонние правила GCP, как и другие материалы ICН, являются межрегиональными, но не носят глобального характера. Отметим, что по числу стран и их площади на государства ICН приходится лишь около 1/10 части земного шара (рисунок), по числу населения – около 15 % жителей планеты.
Документ ВОЗ основан не только на положениях Хельсинской декларации, но также на опубликованных ранее рекомендациях экспертов и разработанных CIOMS международных этических принципах биомедицинских исследований на людях. Соответственно текст ВОЗ признает существующие различия между культурами разных стран. Этот документ направлен на достижение общего понимания и реализации этических принципов во всем мире. Он содержит многие положения, содержащиеся в тексте правил ЕС, в частности, в отношении прав лиц, являющихся объектами исследований, а также ответственности исследователей. В руководстве ВОЗ более детально, чем в других аналогичных документах, отражена специфическая роль органов нормативного контроля лекарств.
Правила GCP ВОЗ отражают сложившуюся в мире практику деления клинических испытаний на фазы; они содержат краткие характеристики четырех фаз. При этом уточняется, что в рамках I и II фаз объектами исследований являются в основном активные ингредиенты (лекарственные субстанции). При описании I фазы отмечается, что она часто проводится на здоровых добровольцах. В отношении III фазы подчеркнуто, что в ее рамках изучаются конкретные лекарственные формы активных ингредиентов, а не сами фармацевтические субстанции.
При составлении текста ICН были учтены правила GCP США, ЕС и ВОЗ. Это руководство имеет целью обеспечить регуляторное признание во всех странах-участницах инициативы документации, содержащей результаты исследований по разработке новых препаратов. Акцент в этом руководстве сделан на обязанностях спонсора, т.е. фирмы-разработчика, что отражает подходы, преобладающие в США и некоторых странах Западной Европы. Правила ICН содержат ссылки на 70 с лишним документов, следование которым необходимо при подготовке и проведении клинических испытаний. Таким образом, сделана попытка охватить все существующие в странах ЕС, США и Японии официальные требования в отношении организации клинических исследований.
Кроме того, следует учитывать, что текст GCP ICН — лишь один из обширного пакета трехсторонних документов, касающихся оценки эффективности новых препаратов. Эти материалы включают большое количество других методических рекомендаций, посвященных различным аспектам организации клинических испытаний. Примеры таких документов приведены в списке литературы [15–17]. В них содержатся весьма существенные разделы в отношении общего порядка работы в данной области, в т. ч. классификация отдельных видов исследований по их целям, а также по фазам. В связи с этим в основном тексте (Е6) классификация опущена. Иначе говоря, помимо самого текста правил GCP для их внедрения необходимо иметь около 80 других документов. По этой причине странам, не входящим в трехстороннюю инициативу, крайне трудно или даже невозможно полностью внедрить Правила ICН.
Терминологический раздел трехстороннего документа расширен в сравнении с соответствующей частью текста ВОЗ. В нем приведены определения двух консультативных органов, призванных обеспечить справедливые экономические и социальные условия участия испытуемых — «Наблюдательный совет учреждения» (Institutional Review Board, IRB) и «Независимый этический комитет» (Independent Ethics Committee, IEC). Их функции и состав весьма близки и создаются они на альтернативной основе (либо один, либо другой). Первый из них отражает опыт США, второй – практику отдельных государств Западной Европы.
Определения двух почти идентичных органов появились в связи с тем, что при согласовании текста американские специалисты не смогли договориться со своими европейскими коллегами относительно одной дефиниции, которая устроила бы всех (включая представителей Японии). Для специалистов стран-участниц ICН соседство двух определений не осложняет пользование документом, поскольку они имеют достаточное представление либо о Совете учреждения, либо об Этическом комитете. Однако в странах, не имеющих еще необходимого опыта в области применения правил GCP, описание двух близких органов в одном нормативном документе без каких-либо пояснений может вызвать недоумение. Положение усугубляется упоминанием третьего аналогичного органа – «Независимого комитета по мониторингу данных» (Independent Data-Monitoring Committee, IDMC). Исходя из этих соображений, в Правилах GCP ВОЗ прописан лишь один орган – «Этический комитет» (еthics сommittee).
Рассматриваемые руководства по GCP по-разному трактуют принцип информированного согласия на участие в испытаниях. В документе ВОЗ, в полном соответствии с Хельсинской декларацией (в формулировке, существовавшей на момент утверждения текста ВОЗ), четко указано, что для участия в нетерапевтических исследованиях, т.е. не приносящих прямой медицинской помощи испытуемым, необходимо письменное согласие добровольца (раздел 3.3.g). Согласно Правилам ICН нетерапевтические исследования могут проводиться также и с согласия представителей лиц-участников (п. 4.8.14). Такая формулировка, безусловно, расширяет возможности экспериментаторов по привлечению дополнительных категорий субъектов исследований. В некоторых культурах, однако, она может повести к ущемлению прав определенных групп лиц. Речь при этом идет не только о малолетних детях или умственно неполноценных гражданах, но и о совершеннолетних и здоровых «младших членах» общины, решения за которых, по традиции, могут принимать глава семьи, лидеры клана и т.п.
Российские правила качественных клинических испытаний
Как уже отмечалось в журнале «Фарматека», советские, а затем и российские авторитеты из числа фармакологов и клиницистов длительное время отвергали принципы Хельсинской декларации и основанные на них Правила GCP [4]. Однако во второй половине 90-х гг., по мере возрастания числа предложений зарубежных фармацевтических фирм о проведении в России испытаний своих препаратов, ситуация начала меняться в лучшую сторону.
В 1996 г., после утверждения окончательного варианта трехсторонних Правил GCP, представители крупнейших фармацевтических корпораций обратились в Минздрав РФ с предложением принять эти Правила в качестве национального стандарта. Такое решение было выгодно, прежде всего, зарубежным фирмам. Есть все основания полагать, что ведущие разработчики и производители лекарств организуют испытания своих препаратов в странах с потенциально емким рынком не столько в научных, сколько в коммерческих целях – для завоевания рынка. Для выполнения этой основной задачи методический уровень проводимых в стране исследований не столь важен; здесь главное – приучить врачей и пациентов к своим «брендам». Кроме того, работа по «гармонизированным» правилам GCP дает возможность фирмам добирать дополнительное число пролеченных пациентов и, тем самым, ускорять регистрацию соответствующих препаратов в странах-участницах ICН. Таким образом, переход отечественных клинических баз на работу по трехсторонним правилам GCP позволяет инофирмам получать двойную выгоду от испытаний своих препаратов в России.
Вместе с тем, в расширении объемов испытаний зарубежных препаратов заинтересована и российская сторона. Благодаря этому в страдающие от нехватки лекарств лечебно-профилактические учреждения бесплатно поступают эффективные медикаменты. Руководители и ведущие специалисты клинических центров получили возможность дополнительно заработать и, одновременно, участвовать в научных исследованиях, публиковать их результаты. Не остается в накладе и Минздрав, поскольку увеличение количества клинических испытаний, проводимых в стране, ведет к росту числа регистрируемых зарубежных препаратов.
К этому моменту окончательно выяснилась неспособность работников Минздрава РФ подготовить отечественные правила GCP, несмотря на щедро раздававшиеся обещания «вот-вот» выпустить такой документ [18, 19]. Таким образом, предложение инофирм пришлось как нельзя более кстати. Возможно, именно поэтому министерские чиновники поспешили согласиться на него, не прочитав внимательно предложенный для утверждения текст. Так появился на свет парадоксальный по форме документ «Правила проведения качественных клинических испытаний в Российской Федерации». Согласно его титульному листу, дословный перевод текста ICН с прямым указанием на то, что он распространяется на страны ЕС, Японию и США*, был одобрен Фармакологическим государственным** комитетом (протокол № 8 от 26 июня 1997 г.), а также утвержден Начальником Управления государственного** контроля лекарственных средств и медтехники (12 августа 1997 г.) и первым заместителем Министра здравоохранения РФ (7 сентября 1997 г.). Иными словами, государственные служащие одобрили документ, не имевший касательства к государству, на службе которого они состояли.
Можно предположить, что в дальнейшем это уникальное творение все же попало на глаза юридически грамотным людям, поскольку год спустя его текст был надлежащим образом отредактирован и оформлен в виде ОСТа 42–511–99 [20]. Следует отметить, что в принятом в 1998 г. Федеральном законе «О лекарственных средствах» нашли отражение многие принципы Хельсинской декларации и, соответственно, правил GCP.
Ошибки в оформлении первых российских правил GCP затронуты здесь в связи с тем, что они свидетельствуют о непонимании многими работниками отечественного фармацевтического сектора механизмов международного сотрудничества в данной области. Взвешенный взгляд на эти механизмы отражен в позиции украинских специалистов: «Для участия в международных кооперативных клинических исследованиях местные регуляторные требования необходимо привести в соответствие с регламентом Международной конференции по гармонизации, учесть рекомендации ВОЗ, Европейского этического комитета и рабочей группы по этике Европейского форума по GCP» [21]. Из всего этого комплекса ориентиров российское медицинское начальство заметило только один.
Очевидно, что в России при выборе основы для принятия национальных требований GCP следовало бы рассмотреть альтернативы. Насколько можно судить, вариант использования руководства ВОЗ не рассматривался. Между тем, имеются веские аргументы в пользу именно такого варианта.
Основное преимущество правил ВОЗ заключается в том, что они пригодны для применения в любом регионе мира и в странах с различными традициями, особенностями культуры и социально-экономическими условиями. Российские специалисты и Минздрав РФ участвовали в разработке и утверждении этого документа на всех этапах: предварительные консультации, заседания экспертного комитета, уставная сессия Организации. Иначе говоря, данное руководство составлено при участии России и адресовано всем странам-членам ВОЗ, в т.ч. и России.
Пояснения относительно деления испытаний на фазы и возможности использования здоровых лиц в качестве испытуемых, не прописанные в основном тексте правил ICН, представляются исключительно важными для России в связи с отсутствием соответствующих указаний* в отечественных нормативных документах. Как отмечено выше, эти пояснения не вошли в текст GCP ICН лишь потому, что они прописаны более детально в других трехсторонних документах, таких как «Общие соображения относительно клинических испытаний» [15] и др.
В официальных российских документах экспериментальные препараты до этапа их регистрации, как правило, именуются «фармакологическими веществами», что соответствует понятию «активные ингредиенты» согласно международной терминологии. Это грубо искажает внутреннюю сущность предрегистрационных клинических испытаний, так как согласно общепринятой практике, отраженной в материалах ВОЗ, объектами наиболее массовых этапов исследований являются не сами лекарственные субстанции (или «фармакологические вещества»), но их специфические дозированные формы.
Именно по этой причине зарегистрировать в так называемых «цивилизованных странах» можно лишь фармацевтический (лекарственный) продукт, т.е. определенную дозированную форму конкретного производителя, в характерной упаковке и произведенную по определенной технологии. Сами лекарственные субстанции в этих странах регистрации не подлежат. Российские же регуляторные органы до сего времени не уловили принципиального различия между активными субстанциями и фармацевтическими продуктами, в связи с чем и допускают регистрацию субстанций.
В пользу текста ICН говорит то соображение, что им пользуются практически все ведущие в мире фирмы-разработчики новых препаратов, на долю которых приходится свыше 80 % выводимых на мировой рынок оригинальных лекарств. По этой причине принятие его в России может содействовать дальнейшему расширению участия отечественных клинических баз в международных многоцентровых исследованиях лекарственных средств. При этом, однако, следует учитывать тот факт, что руководство ICН по GCP отражает уровень организации клинических испытаний, еще не достигнутый в нашей стране. Как уже отмечалось, в России официально не признана практика деления клинических испытаний на фазы, нет полной ясности относительно возможности проведения отдельных этапов исследований на здоровых людях. Этические комитеты учреждений только начинают создаваться; в этом деле остается много спорного [23]. Отсутствуют десятки нормативов, необходимых для внедрения трехсторонних правил GCP. В странах-участницах Международной конференции по гармонизации эти базовые условия были достигнуты уже 10–20 лет назад. По этой причине в рамках трехсторонней инициативы основное внимание теперь сконцентрировано не на том, как правильно проводить клинические испытания, а на унификации порядка документального оформления результатов исследований с тем, чтобы сократить и упростить процедуру регистрации лекарств в странах-участницах ICН.
К тому же российские специалисты и учреждения не могли влиять на содержание указанного документа, поскольку, как известно, Россия не входит в число стран-участниц ICН. Иными словами, текст ICН был подготовлен без нашего участия и не учитывает особенностей ситуации в России. В свете вышесказанного утверждения о том, что отраслевой стандарт «Правила проведения качественных клинических испытаний в Российской Федерации» максимально приближен к рекомендациям ВОЗ [24] не представляются вполне объективными.
В то же время это не означает, что решение о признании руководства ICН в принципе невыгодно для российской стороны. Выгода, причем не только сиюминутная, но и в стратегическом плане, может быть получена, однако она не придет автоматически. Для ее извлечения необходимо в кратчайший срок сократить существующий в настоящее время разрыв в уровне проведения клинических испытаний между Россией, с одной стороны, и США, Великобританией и Швецией – с другой. С этой целью необходимо незамедлительно разработать и утвердить внушительный пакет нормативов в поддержку правил GCP. Один норматив этого ряда – специфические правила GMP, применимые к производству предназначенных для проведения клинических испытаний экспериментальных препаратов, хотелось бы рассмотреть подробнее.
Правила GMP для производства экспериментальных препаратов
Оба рассматриваемых руководства по GCP указывают на необходимость изготовления исследуемых препаратов в соответствии с принципами GMP (п. 5.6 текста ВОЗ; п. 5.13.1 текста ICН). Такое же требование содержится в проекте Директивы ЕС (2001) по клиническим испытаниям (п. 12). Еще в январе 1991 г. в документе Европейского Сообщества (111/3044/91), опубликованном для обсуждения, было признано нелогичным, что экспериментальные препараты не подлежат контролю, обязательному в отношении тех лекарственных средств, прототипами для которых они являются. Большинство комментариев на документ, поступивших от заинтересованных сторон, подтверждало это мнение.
Однако экспериментальные препараты, особенно используемые на ранних этапах клинического изучения, обычно производятся не по утвержденному регламенту, часто в отсутствие окончательных данных о свойствах дозированной формы, а иногда и активной субстанции. Поэтому правила GMP не могут быть применены к ним в полном объеме. Следовательно, необходимы поправки и уточнения на этот счет. С учетом этих соображений, все 3 широко признанные в мире кодекса GMP (ВОЗ, ЕС и PIC) содержат приложения, уточняющие специальные требования к производству экспериментальных продуктов.
Приложения к правилам GMP для производства исследуемых образцов касаются тех аспектов GMP, которые в отношении этой категории препаратов могут иметь свои особенности. Они также включают указания относительно предписания к выдаче, отгрузки и возврата клинических запасов, которые увязаны с правилами GCP.
В соответствующем тексте ВОЗ [25] отмечается: «Настоящее руководство является дополнением как к руководству по GMP, так и руководству по надлежащей клинической практике (GCP) испытаний фармацевтических препаратов. Применение принципов GMP к изготовлению исследуемых продуктов необходимо по нескольким соображениям:
Чтобы гарантировать постоянство между сериями и внутри серий исследуемого препарата и обеспечить, таким образом, надежность клинических испытаний. Чтобы гарантировать соответствие свойств исследуемых образцов и препаратов будущего промышленного изготовления и, тем самым, обеспечить применимость клинического испытания к эффективности и безопасности препарата, который поступит на рынок. Чтобы защитить субъектов клинических испытаний от некачественных продуктов, полученных в результате производственных ошибок (пропуск таких критических стадий, как стерилизация, контаминация и перекрестная контаминация, пересортица, ошибочная маркировка и т.д.) или из-за низкого качества исходного сырья и материалов первичной упаковки». Как следует из материалов ВОЗ, крайне важен выбор дозированной формы для клинических испытаний. Известно, что на ранних этапах исследований лекарственная форма может существенным образом отличаться от окончательной. Например, в исследованиях препарат может приниматься в капсулах, в то время как коммерческий продукт намечено выпускать в форме таблеток. Однако в III фазе испытаний лекформа должна быть такой же, как планируемая для промышленного производства. В противном случае результаты клинических испытаний не будут надежно удостоверять эффективность и безопасность препарата, поступающего на рынок.
Если есть значительное различие в лекарственных формах на этапе клинических испытаний и при выходе на рынок, необходимо представить в регистрационные органы данные, подтверждающие, что окончательная лекарственная форма эквивалентна в части биодоступности и стабильности той, что использовалась при клинических испытаниях. После внесения изменений в пропись лекформы и в технологические процессы, вслед за масштабированием и переносом производства на другие участки, окончательно выбранные производственные условия должны пройти ревалидацию.
Традиционным подходом к изготовлению «клинических образцов» является использование опытно-наработочной, иначе пилотной, установки (pilot plant), предназначенной, в первую очередь, для отработки технологии. Такой подход экономически наиболее выгоден предприятиям, поскольку избавляет их от необходимости создавать специальные производственные линии. К тому же продукция, получаемая в результате технологических экспериментов, не списывается, но используется в медико-биологических исследованиях. Однако такое совмещение противоречит принципам GMP, в частности, требованию постоянства условий производства одного продукта от серии к серии. Очевидно, что отработка технологии невозможна без изменения процессов, а иногда и прописи лекформы.
Соответственно, по мере внедрения правил GMP в фармацевтическое производство, стали использоваться другие варианты изготовления клинических материалов. Среди них можно выделить следующие:
-
Установка, смонтированная только для выпуска экспериментальных препаратов и не предназначенная для отработки технологии. При этом нередко используется малогабаритная аппаратура, аналогичная по конструкции и принципам действия полномасштабному промышленному оборудованию. На некоторых фирмах такие установки неофициально именуются «аптеками» (pharmacy). Очевидно, такое название призвано подчеркнуть необходимость соблюдения чистоты, порядка и аккуратности при работе с ними.
-
Производственная линия, предназначенная для выпуска как образцов для III фазы испытаний, так и для последующего промышленного производства коммерческой продукции в начальный период времени (например, 1–2 года) после регистрации препарата (так называемое «первичное производство»).
Такие варианты организации производства клинических образцов не противоречат принципам GMP. В тех случаях, когда предполагается достаточно большой расход «образцов для клиники», например, при международных многоцентровых исследованиях, практикуется их выпуск в условиях полномасштабного производства.
Некоторые производственные процессы для исследуемых лекарственных средств могут не проходить валидацию в таком объеме, который требуется для серийного производства. Иногда нет необходимости разрабатывать регламент и технологические инструкции, но для каждой производственной операции или поставки должны составляться четкие письменные инструкции и протоколы. Протоколы имеют особенное значение для подготовки окончательной версии документации, которая будет использоваться в серийном производстве. Протоколы серий должны храниться по меньшей мере 2 года после завершения клинического испытания.
Предписание может содержать требование приготовления и/или упаковки определенного количества единиц, а также их транспортировки. Только спонсор может выдать предписание производителю исследуемых препаратов. Оно должно представляться в письменном виде и быть достаточно точным, чтобы избежать ошибок в толковании. Предписание должно быть официально санкционировано и относится к утвержденному досье спецификаций на препарат.
Вся информация, необходимая для составления подробных письменных инструкций по изготовлению, упаковке, контролю качества, реализации серии, условиям хранения и транспортирования, должна содержаться в досье спецификаций на препарат. Досье необходимо постоянно обновлять, обеспечивая при этом прослеживаемость предыдущих версий.
Спецификации могут изменяться в процессе разработки препарата. Эти изменения должны вноситься в соответствии с письменной методикой, утвержденной ответственным лицом, и четко протоколироваться. Спецификации должны основываться на всех имеющихся научных данных, современном уровне науки и техники, применяемой технологии, а также на фармакопейных и других официальных требованиях.
Пропись и технологические инструкции могут изменяться на основании достигнутого опыта. Необходимо, однако, учитывать все возможные последствия для стабильности и, особенно, биоэквивалентности отдельных серий готовой продукции. Изменения должны вноситься в соответствии с письменной методикой, утвержденной ответственным лицом, и четко протоколироваться.
Операции по упаковке и маркировке исследуемых препаратов с использованием закодированных «слепых» этикеток в большей степени подвержены ошибкам, чем упаковка и маркировка зарегистрированных препаратов. Следовательно, необходимо усилить процедуры надзора за соответствием этикеток, чистотой линии и т.д.
Инструкции по упаковке основываются на предписании. Серии исследуемых образцов могут быть подразделены на различные партии для упаковки. Количество образцов, подлежащих упаковке, должно быть определено до начала операций по упаковке, при этом необходимо учесть количество образцов, необходимых для проведения контроля качества, и тех, которые должны быть заложены на хранение. По окончании процесса упаковки и маркировки необходимо составить баланс.
Этикетки экспериментальных препаратов должны содержать название (имя) организатора (спонсора), код испытания, номер серии, идентификационный номер пациента, условия хранения и срок годности с указанием месяца/года или даты повторного испытания.
В соответствии с предписанием, на этикетках может быть размещена дополнительная информация. При необходимости, в условиях слепого исследования, номер серии может указываться отдельно. Копия каждого типа этикеток должна сохраняться в протоколе упаковки серии.
Протоколы производства и упаковки серии должны содержать достаточно подробную информацию для точного прослеживания всех операций. Эти протоколы должны включать все относящиеся к делу замечания, которые расширяют существующие знания о препарате, позволяют усовершенствовать производственные операции и обосновать используемые методики. Необходимы доказательства того, что эти изменения не повлияют на первоначальные качественные характеристики препарата.
Специальные методики должны описывать создание, распределение, обработку и сохранение любого кода рандомизации, использованного для упакованных исследуемых препаратов. Должна применяться система кодирования, позволяющая точно идентифицировать закодированную («слепую») продукцию. Этот код, вместе с кодом рандомизации и списком рандомизации, должен позволять точно идентифицировать препарат, включая любую необходимую прослеживаемость кодов и номера серии препарата. Необходимо сохранять образцы закодированных исследуемых препаратов.
Ответственным за уничтожение неиспользованных исследуемых препаратов является не производитель, а спонсор. Таким образом, исследуемые препараты не могут быть уничтожены производителем без предварительной санкции организатора испытания.
Глобальная гармонизация руководящих принципов в отношении клинических исследований
В настоящее время во многих странах работники фармацевтического сектора стоят перед нелегким выбором: ориентироваться на глобальные нормы и правила ВОЗ или внедрять руководства ICН без учета достигнутого уровня развития здравоохранения и экономики? В сложившейся ситуации Секретариат ВОЗ проводит интенсивную работу по поиску консенсуса, сближению требований и методических материалов, разрабатываемых в рамках Организации и через механизмы межрегионального сотрудничества. Это касается широкого круга нормативов, в т.ч. правил GCP.
Как уже отмечалось, текст Хельсинской декларации, лежащей в основе этих правил, неоднократно пересматривался [4]. Особенно значительные поправки были внесены в него в конце 2000 г. на 52 сессии Всемирной медицинской ассоциации, состоявшейся в Эдинбурге. Из 32 статей Декларации лишь 3 остались неизмененными, а 8 являются новыми. Изменения касаются ограничений в использовании плацебо, получения терапевтической пользы участниками испытаний, обеспечения прозрачности финансовой поддержки исследований и ряда других важных положений. Пересматриваются также и этические принципы биомедицинских исследований на людях, опубликованные CIOMS в 1993 г. и конкретизирующие многие положения Хельсинской декларации.
В последние годы мировое общественное здравоохранение столкнулось с новыми вызовами в виде опасных заболеваний, таких как ВИЧ-инфекция и гепатит С. В этих условиях требуются новые подходы к оценке соотношения польза/риск терапевтических вмешательств. Наряду с этим, возрастает потребность в проведении клинических исследований на детях, пожилых людях, во время беременности, в условиях вертикальной (от матери к новорожденному) передачи ВИЧ-инфекции [26]. Существует также объективная необходимость разработки принципов GCP для испытания вакцин и других биологических препаратов, имеющих существенные отличия от химико-фармацевтическ